Department of Biochemistry, Genetics, and Immunology, University of Vigo, Vigo, Spain.
Genome Biol Evol. 2011;3:896-908. doi: 10.1093/gbe/evr080. Epub 2011 Aug 7.
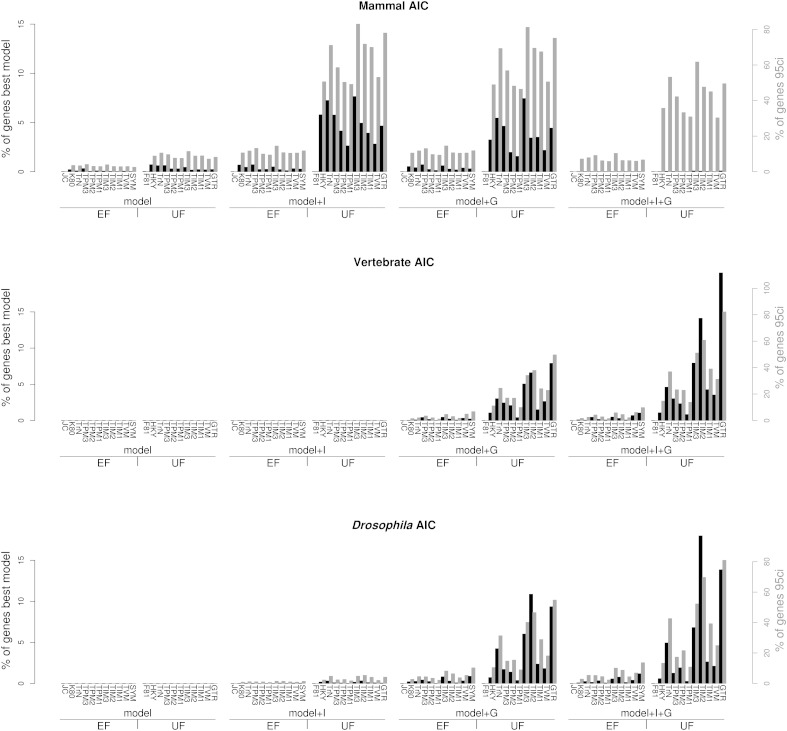
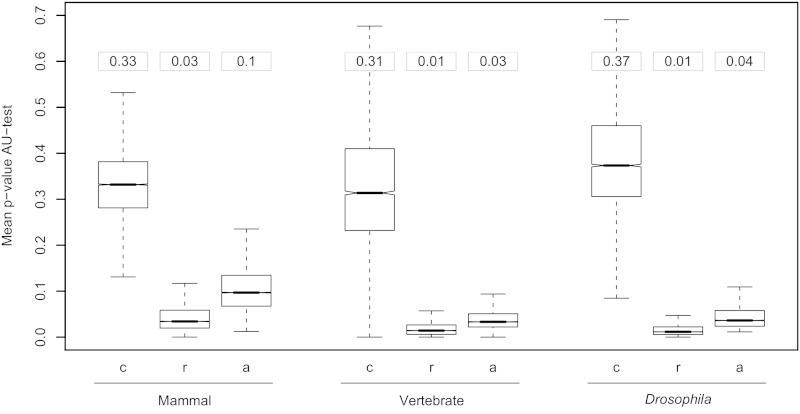
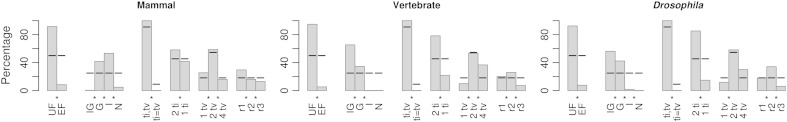
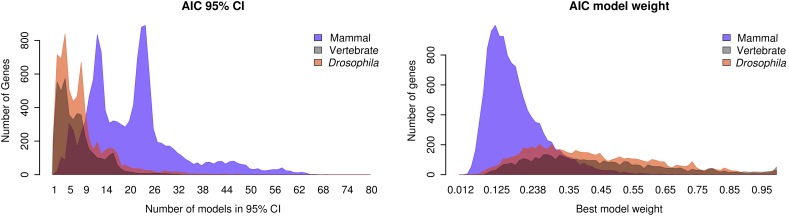
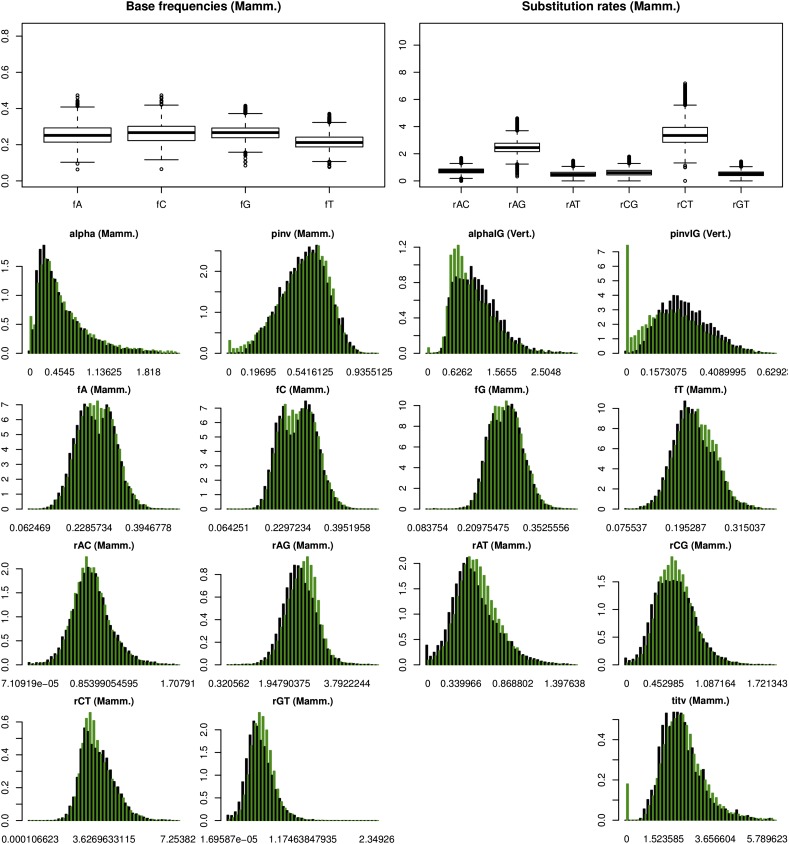
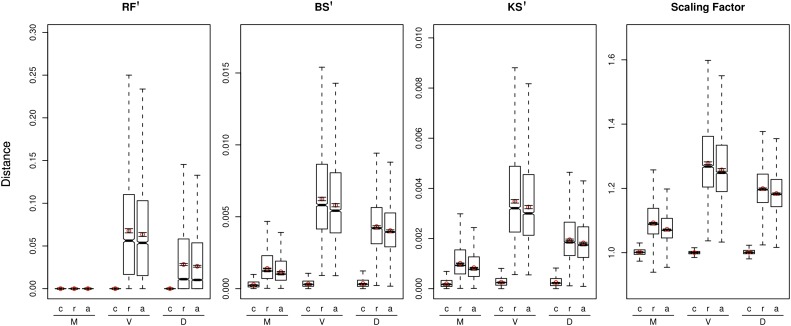
At a genomic scale, the patterns that have shaped molecular evolution are believed to be largely heterogeneous. Consequently, comparative analyses should use appropriate probabilistic substitution models that capture the main features under which different genomic regions have evolved. While efforts have concentrated in the development and understanding of model selection techniques, no descriptions of overall relative substitution model fit at the genome level have been reported. Here, we provide a characterization of best-fit substitution models across three genomic data sets including coding regions from mammals, vertebrates, and Drosophila (24,000 alignments). According to the Akaike Information Criterion (AIC), 82 of 88 models considered were selected as best-fit models at least in one occasion, although with very different frequencies. Most parameter estimates also varied broadly among genes. Patterns found for vertebrates and Drosophila were quite similar and often more complex than those found in mammals. Phylogenetic trees derived from models in the 95% confidence interval set showed much less variance and were significantly closer to the tree estimated under the best-fit model than trees derived from models outside this interval. Although alternative criteria selected simpler models than the AIC, they suggested similar patterns. All together our results show that at a genomic scale, different gene alignments for the same set of taxa are best explained by a large variety of different substitution models and that model choice has implications on different parameter estimates including the inferred phylogenetic trees. After taking into account the differences related to sample size, our results suggest a noticeable diversity in the underlying evolutionary process. All together, we conclude that the use of model selection techniques is important to obtain consistent phylogenetic estimates from real data at a genomic scale.
在基因组尺度上,塑造分子进化的模式被认为是高度异质的。因此,比较分析应该使用适当的概率替代模型,这些模型可以捕捉不同基因组区域进化的主要特征。虽然人们集中精力开发和理解模型选择技术,但目前还没有报道过在基因组水平上对总体相对替代模型拟合的描述。在这里,我们对三个基因组数据集(包括哺乳动物、脊椎动物和果蝇的编码区的 24,000 个比对)中的最佳替代模型进行了特征描述。根据赤池信息量准则(AIC),在所考虑的 88 个模型中,有 82 个模型至少在一次情况下被选为最佳拟合模型,尽管频率差异非常大。大多数参数估计值在基因之间也有很大的差异。在脊椎动物和果蝇中发现的模式与在哺乳动物中发现的模式非常相似,而且往往更复杂。从置信区间集内的模型得出的系统发育树显示出的方差要小得多,并且比从置信区间外的模型得出的树与最佳拟合模型估计的树更接近。虽然替代标准选择的模型比 AIC 更简单,但它们也提出了类似的模式。总之,我们的研究结果表明,在基因组尺度上,同一组分类群的不同基因比对最好用大量不同的替代模型来解释,并且模型选择对包括推断的系统发育树在内的不同参数估计值有影响。在考虑到样本大小差异后,我们的研究结果表明,在潜在的进化过程中存在明显的多样性。总之,我们的结论是,在基因组尺度上,使用模型选择技术对于从真实数据中获得一致的系统发育估计是很重要的。