Department of Biochemistry, Center for Structural Biology, Vanderbilt University School of Medicine, Nashville, TN 37232-8725, USA.
Structure. 2011 Aug 10;19(8):1160-9. doi: 10.1016/j.str.2011.05.009.
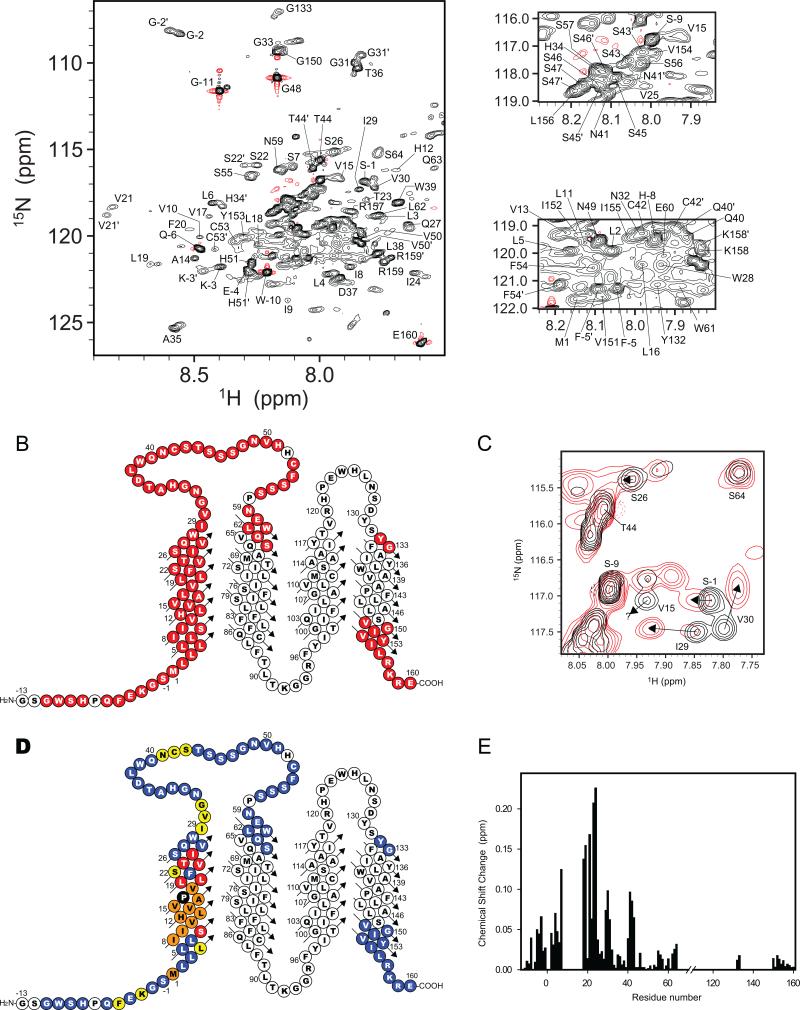
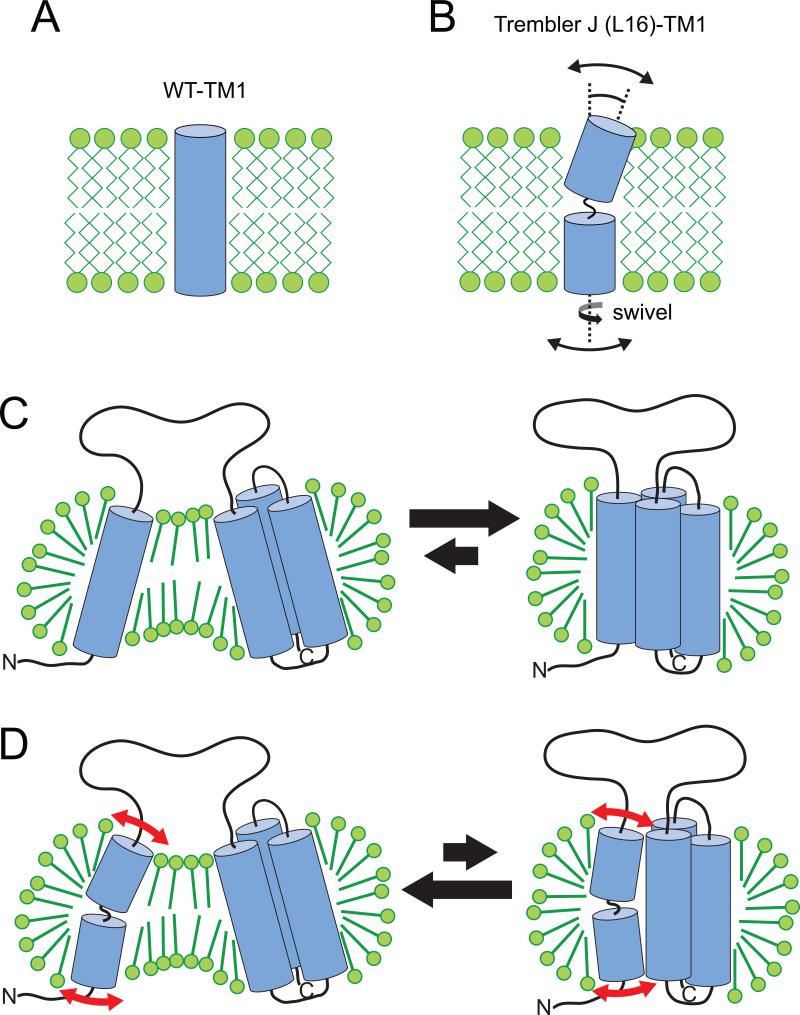
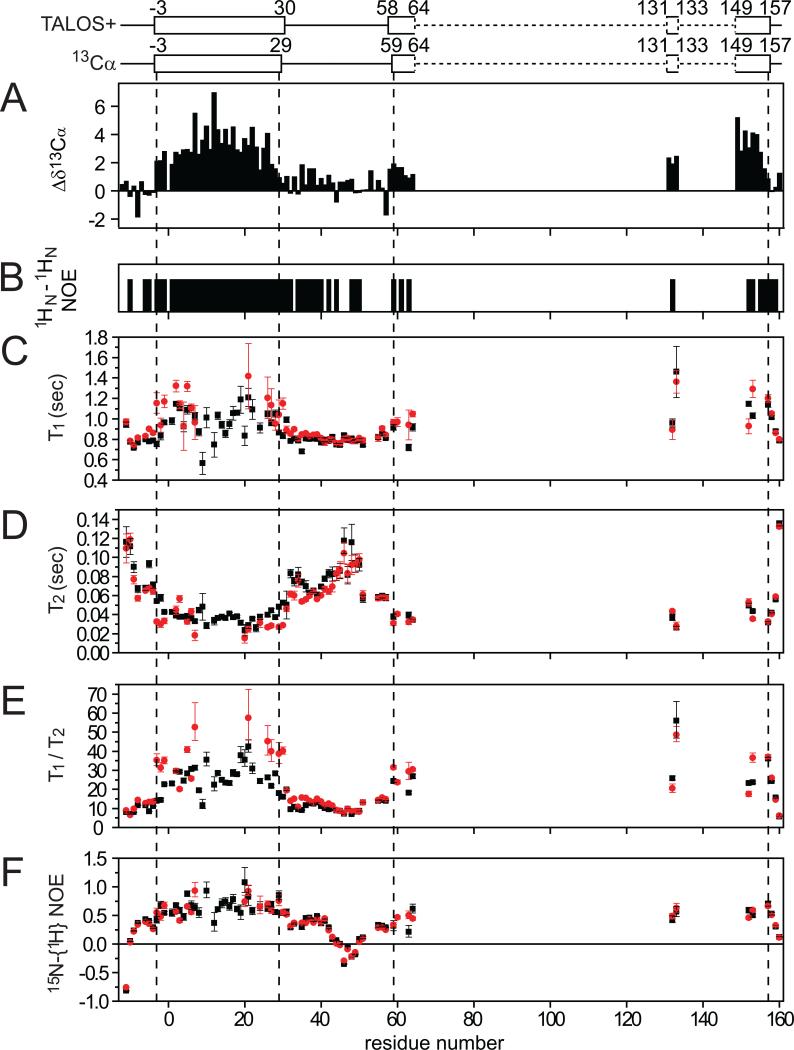
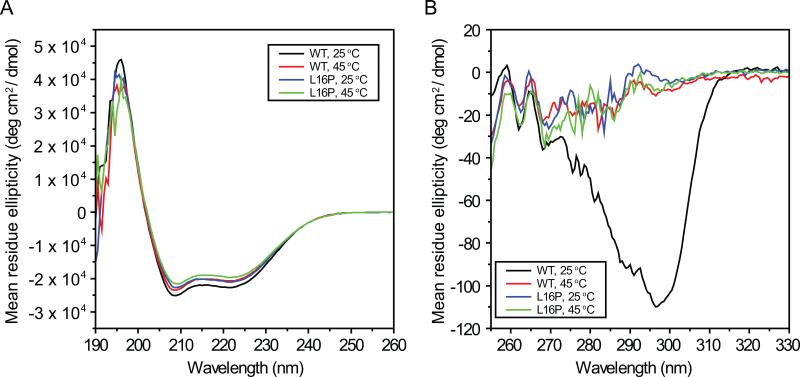
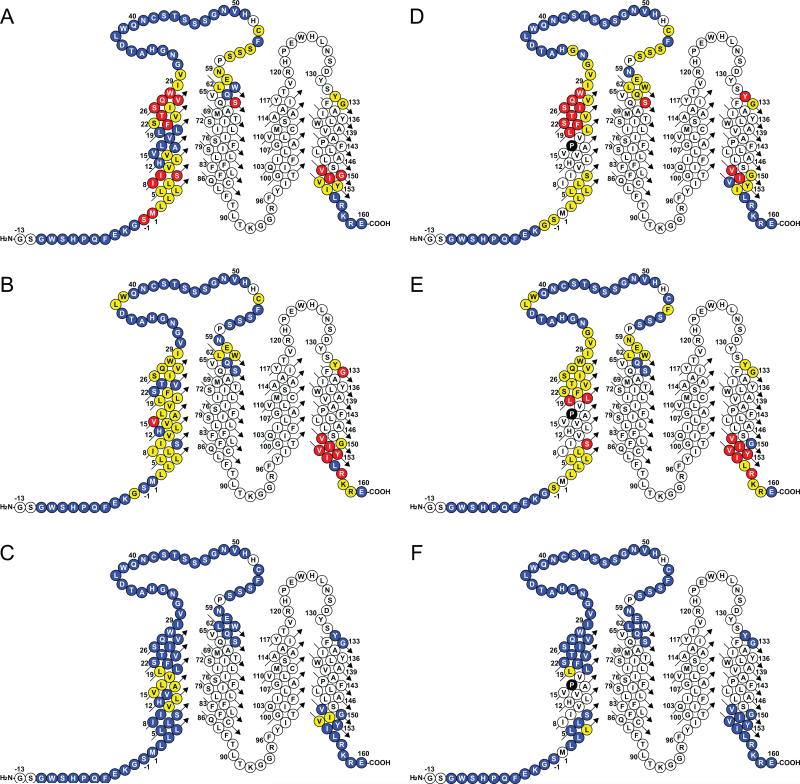
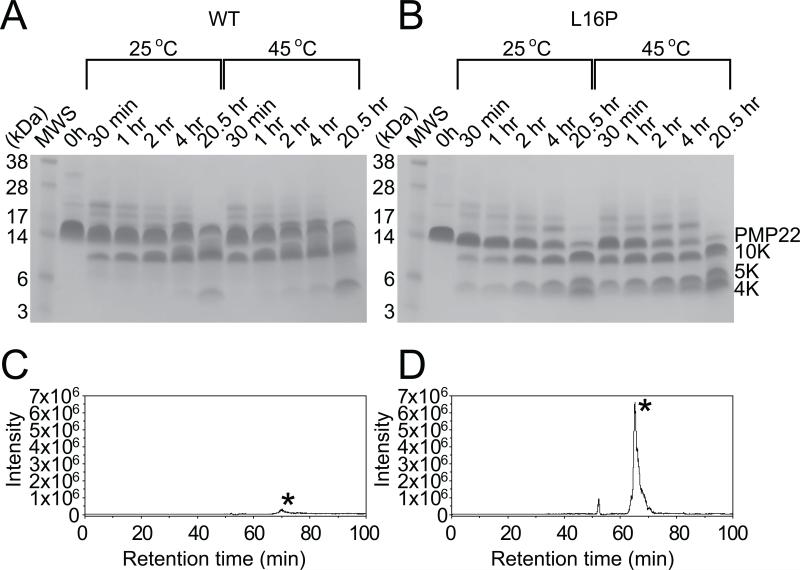
Mutations in peripheral myelin protein 22 (PMP22) can result in the common peripheral neuropathy Charcot-Marie-Tooth disease (CMTD). The Leu16Pro mutation in PMP22 results in misassembly of the protein, which causes the Trembler-J (TrJ) disease phenotype. Here we elucidate the structural defects present in a partially folded state of TrJ PMP22 that are decisive in promoting CMTD-causing misfolding. In this state, transmembrane helices 2-4 (TM2-4) form a molten globular bundle, while transmembrane helix 1 (TM1) is dissociated from this bundle. The TrJ mutation was seen to profoundly disrupt the TM1 helix, resulting in increased backbone dynamics and changes in the tertiary interactions of TM1 with the PMP22 TM2-4 core in the folded state. Consequently, TM1 undergoes enhanced dissociation from the other transmembrane segments in TrJ PMP22, becoming available for recognition and sequestration by protein-folding quality control, which leads to loss of function and toxic accumulation of aggregates that result in CMTD.
外周髓鞘蛋白 22(PMP22)的突变可导致常见的周围神经病变——遗传性运动感觉神经病(CMTD)。PMP22 中的亮氨酸 16 脯氨酸突变导致蛋白组装错误,从而引起 Trembler-J(TrJ)疾病表型。在这里,我们阐明了 TrJ PMP22 部分折叠状态下存在的结构缺陷,这些缺陷对于促进 CMTD 引起的错误折叠是决定性的。在这种状态下,跨膜螺旋 2-4(TM2-4)形成一个熔融球蛋白束,而跨膜螺旋 1(TM1)与该束解离。TrJ 突变被认为严重破坏了 TM1 螺旋,导致其骨架动力学增加,并改变了 TM1 与折叠状态下 PMP22 TM2-4 核心的三级相互作用。因此,TM1 在 TrJ PMP22 中更容易与其他跨膜片段解离,从而更容易被蛋白质折叠质量控制识别和隔离,这导致功能丧失和有毒聚集物的积累,从而导致 CMTD。