Department of Pathology, University of Arizona, Tucson, Arizona, United States of America.
PLoS One. 2011;6(8):e22267. doi: 10.1371/journal.pone.0022267. Epub 2011 Aug 4.
Gene expression profiling yields quantitative data on gene expression used to create prognostic models that accurately predict patient outcome in diffuse large B cell lymphoma (DLBCL). Often, data are analyzed with genes classified by whether they fall above or below the median expression level. We sought to determine whether examining multiple cut-points might be a more powerful technique to investigate the association of gene expression with outcome.
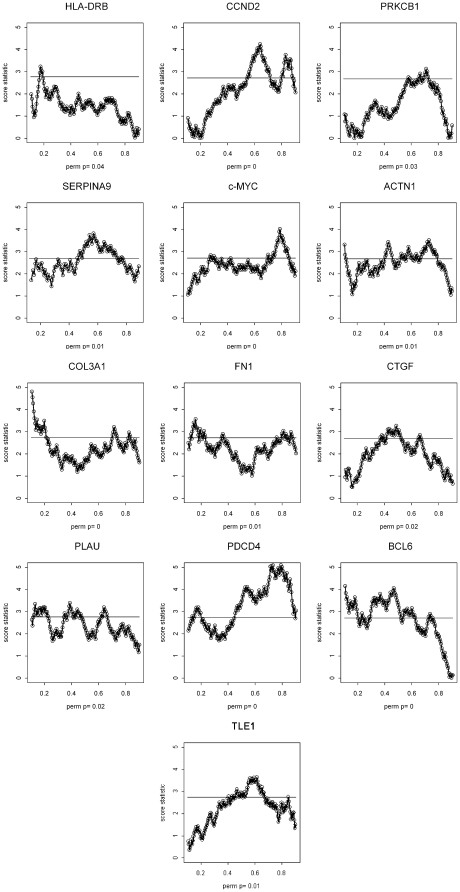
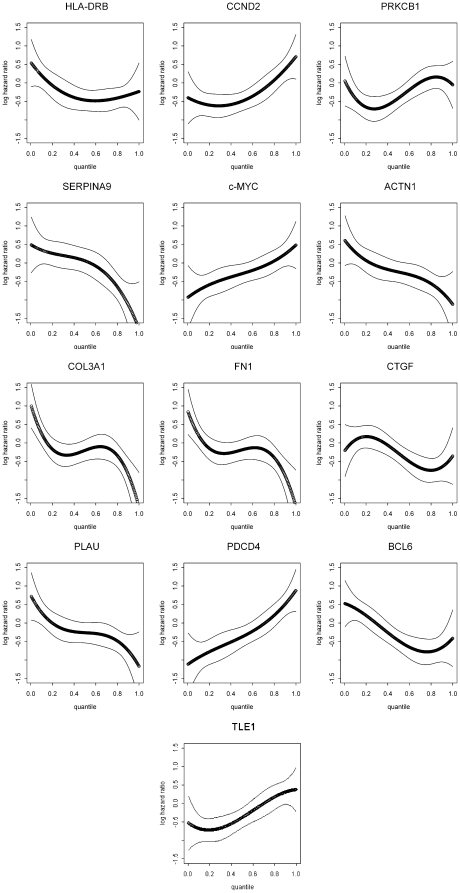
METHODOLOGY/PRINCIPAL FINDINGS: We explored gene expression profiling data using variable cut-point analysis for 36 genes with reported prognostic value in DLBCL. We plotted two-group survival logrank test statistics against corresponding cut-points of the gene expression levels and smooth estimates of the hazard ratio of death versus gene expression levels. To facilitate comparisons we also standardized the expression of each of the genes by the fraction of patients that would be identified by any cut-point. A multiple comparison adjusted permutation p-value identified 3 different patterns of significance: 1) genes with significant cut-point points below the median, whose loss is associated with poor outcome (e.g. HLA-DR); 2) genes with significant cut-points above the median, whose over-expression is associated with poor outcome (e.g. CCND2); and 3) genes with significant cut-points on either side of the median, (e.g. extracellular molecules such as FN1).
CONCLUSIONS/SIGNIFICANCE: Variable cut-point analysis with permutation p-value calculation can be used to identify significant genes that would not otherwise be identified with median cut-points and may suggest biological patterns of gene effects.
基因表达谱可提供用于创建预后模型的基因表达的定量数据,这些模型可准确预测弥漫性大 B 细胞淋巴瘤 (DLBCL) 患者的结局。通常,通过将基因分类为高于或低于中位数表达水平来分析数据。我们试图确定检查多个截断值是否是一种更强大的技术,可以研究基因表达与结局的关联。
方法/主要发现:我们使用可变截断分析探索了 36 个具有 DLBCL 预后价值的基因的基因表达谱数据。我们将两群组生存对数秩检验统计量与相应的基因表达水平的截断值以及死亡与基因表达水平的风险比的平滑估计值进行对比。为了便于比较,我们还通过任何截断值识别出的患者比例来标准化每个基因的表达。多重比较调整的置换 p 值确定了 3 种不同的显著模式:1)具有显著截断值低于中位数的基因,其丢失与不良结局相关(例如 HLA-DR);2)具有显著截断值高于中位数的基因,其过表达与不良结局相关(例如 CCND2);3)具有显著截断值在中位数两侧的基因,(例如细胞外分子如 FN1)。
结论/意义:具有置换 p 值计算的可变截断分析可用于识别具有中位数截断值否则无法识别的显著基因,并可能提示基因效应的生物学模式。