Colosimo Marc E, Peterson Matthew W, Mardis Scott, Hirschman Lynette
The MITRE Corporation, 202 Burlington Rd, Bedford MA 01730, USA.
Source Code Biol Med. 2011 Aug 18;6:13. doi: 10.1186/1751-0473-6-13.
Current sequencing technology makes it practical to sequence many samples of a given organism, raising new challenges for the processing and interpretation of large genomics data sets with associated metadata. Traditional computational phylogenetic methods are ideal for studying the evolution of gene/protein families and using those to infer the evolution of an organism, but are less than ideal for the study of the whole organism mainly due to the presence of insertions/deletions/rearrangements. These methods provide the researcher with the ability to group a set of samples into distinct genotypic groups based on sequence similarity, which can then be associated with metadata, such as host information, pathogenicity, and time or location of occurrence. Genotyping is critical to understanding, at a genomic level, the origin and spread of infectious diseases. Increasingly, genotyping is coming into use for disease surveillance activities, as well as for microbial forensics. The classic genotyping approach has been based on phylogenetic analysis, starting with a multiple sequence alignment. Genotypes are then established by expert examination of phylogenetic trees. However, these traditional single-processor methods are suboptimal for rapidly growing sequence datasets being generated by next-generation DNA sequencing machines, because they increase in computational complexity quickly with the number of sequences.
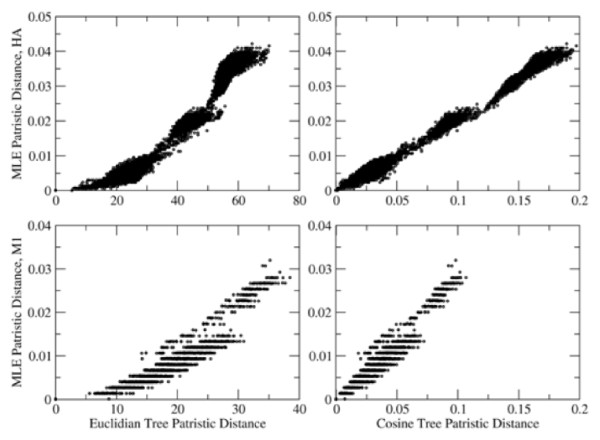
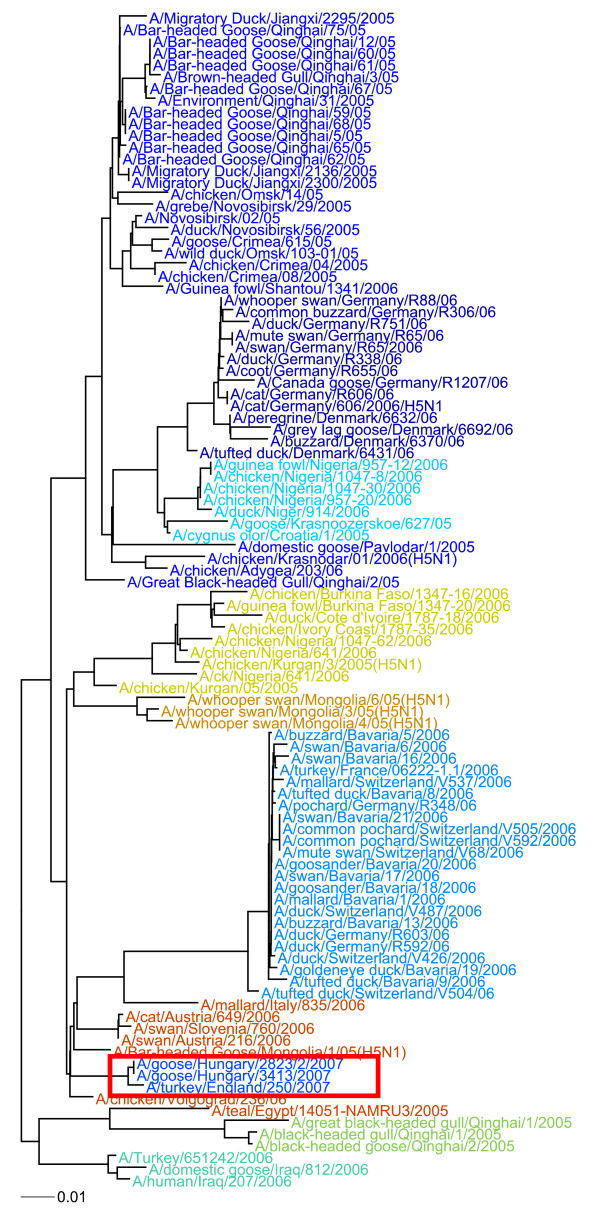
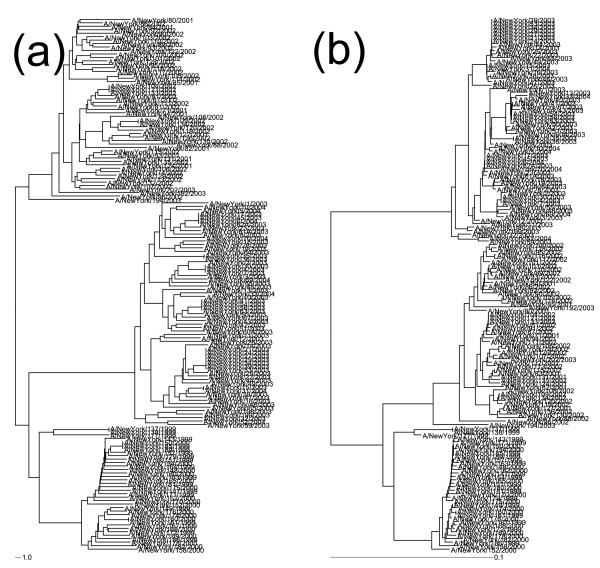
Nephele is a suite of tools that uses the complete composition vector algorithm to represent each sequence in the dataset as a vector derived from its constituent k-mers by passing the need for multiple sequence alignment, and affinity propagation clustering to group the sequences into genotypes based on a distance measure over the vectors. Our methods produce results that correlate well with expert-defined clades or genotypes, at a fraction of the computational cost of traditional phylogenetic methods run on traditional hardware. Nephele can use the open-source Hadoop implementation of MapReduce to parallelize execution using multiple compute nodes. We were able to generate a neighbour-joined tree of over 10,000 16S samples in less than 2 hours.
We conclude that using Nephele can substantially decrease the processing time required for generating genotype trees of tens to hundreds of organisms at genome scale sequence coverage.
当前的测序技术使得对给定生物体的多个样本进行测序成为现实,这给处理和解释带有相关元数据的大型基因组数据集带来了新的挑战。传统的计算系统发育方法对于研究基因/蛋白质家族的进化以及利用这些来推断生物体的进化是理想的,但对于整个生物体的研究并不理想,主要原因是存在插入/缺失/重排。这些方法使研究人员能够根据序列相似性将一组样本分组到不同的基因型组中,然后可以将其与元数据相关联,例如宿主信息、致病性以及发生的时间或地点。基因分型对于在基因组水平上理解传染病的起源和传播至关重要。基因分型越来越多地用于疾病监测活动以及微生物法医学。经典的基因分型方法基于系统发育分析,从多序列比对开始。然后通过专家检查系统发育树来确定基因型。然而,这些传统的单处理器方法对于由下一代DNA测序机器生成的快速增长的序列数据集来说并不理想,因为它们的计算复杂度会随着序列数量的增加而迅速增加。
Nephele是一套工具,它使用完整组成向量算法,通过避免多序列比对的需求,将数据集中的每个序列表示为从其组成的k-mer衍生而来的向量,并使用亲和传播聚类基于向量上的距离度量将序列分组为基因型。我们的方法产生的结果与专家定义的进化枝或基因型高度相关,而计算成本仅为在传统硬件上运行的传统系统发育方法的一小部分。Nephele可以使用MapReduce的开源Hadoop实现,通过多个计算节点并行执行。我们能够在不到2小时的时间内生成超过10000个16S样本的邻接树。
我们得出结论,使用Nephele可以大幅减少在基因组规模序列覆盖下生成数十至数百个生物体的基因型树所需的处理时间。