Graduate Program in Bioinformatics and Medical Informatics, San Diego State University, San Diego, CA, USA.
Center for Microbial Genetics and Genomics, Northern Arizona University, Flagstaff, AZ, USA.
Microbiome. 2016 Feb 24;4:11. doi: 10.1186/s40168-016-0153-6.
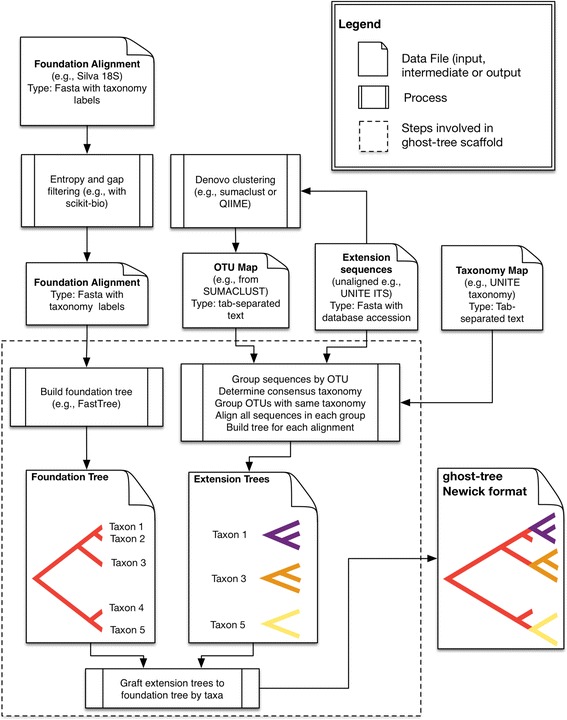
Fungi play critical roles in many ecosystems, cause serious diseases in plants and animals, and pose significant threats to human health and structural integrity problems in built environments. While most fungal diversity remains unknown, the development of PCR primers for the internal transcribed spacer (ITS) combined with next-generation sequencing has substantially improved our ability to profile fungal microbial diversity. Although the high sequence variability in the ITS region facilitates more accurate species identification, it also makes multiple sequence alignment and phylogenetic analysis unreliable across evolutionarily distant fungi because the sequences are hard to align accurately. To address this issue, we created ghost-tree, a bioinformatics tool that integrates sequence data from two genetic markers into a single phylogenetic tree that can be used for diversity analyses. Our approach starts with a "foundation" phylogeny based on one genetic marker whose sequences can be aligned across organisms spanning divergent taxonomic groups (e.g., fungal families). Then, "extension" phylogenies are built for more closely related organisms (e.g., fungal species or strains) using a second more rapidly evolving genetic marker. These smaller phylogenies are then grafted onto the foundation tree by mapping taxonomic names such that each corresponding foundation-tree tip would branch into its new "extension tree" child.
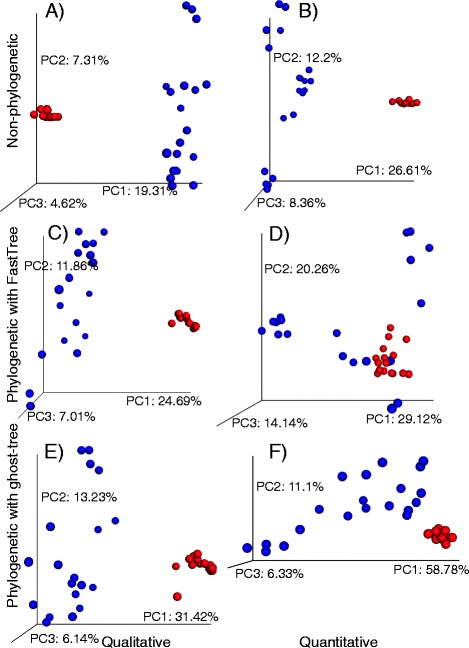
We applied ghost-tree to graft fungal extension phylogenies derived from ITS sequences onto a foundation phylogeny derived from fungal 18S sequences. Our analysis of simulated and real fungal ITS data sets found that phylogenetic distances between fungal communities computed using ghost-tree phylogenies explained significantly more variance than non-phylogenetic distances. The phylogenetic metrics also improved our ability to distinguish small differences (effect sizes) between microbial communities, though results were similar to non-phylogenetic methods for larger effect sizes.
The Silva/UNITE-based ghost tree presented here can be easily integrated into existing fungal analysis pipelines to enhance the resolution of fungal community differences and improve understanding of these communities in built environments. The ghost-tree software package can also be used to develop phylogenetic trees for other marker gene sets that afford different taxonomic resolution, or for bridging genome trees with amplicon trees.
ghost-tree is pip-installable. All source code, documentation, and test code are available under the BSD license at https://github.com/JTFouquier/ghost-tree .
真菌在许多生态系统中起着关键作用,它们会导致动植物患上严重疾病,并对人类健康和建筑环境的结构完整性造成重大威胁。虽然大多数真菌多样性仍不为人知,但聚合酶链式反应(PCR)引物与下一代测序技术相结合,极大地提高了我们对真菌微生物多样性进行分析的能力。虽然内部转录间隔区(ITS)的高序列变异性有助于更准确地鉴定物种,但它也使得跨进化距离较远的真菌进行多序列比对和系统发育分析变得不可靠,因为这些序列难以准确对齐。为了解决这个问题,我们创建了 ghost-tree,这是一种生物信息学工具,它可以将来自两个遗传标记的序列数据整合到一个可以用于多样性分析的单一系统发育树中。我们的方法首先从一个遗传标记开始构建一个“基础”系统发育树,该标记的序列可以在跨越不同分类群的生物体(例如真菌科)中进行对齐。然后,使用第二个进化更快的遗传标记为更密切相关的生物体(例如真菌种或菌株)构建“扩展”系统发育树。然后,通过映射分类名称,将这些较小的系统发育树“嫁接”到基础树上,使得每个对应的基础树末梢都会分支成新的“扩展树”子树。
我们将 ghost-tree 应用于将源自 ITS 序列的真菌扩展系统发育树嫁接在源自真菌 18S 序列的基础系统发育树上。我们对模拟和真实真菌 ITS 数据集的分析发现,使用 ghost-tree 系统发育树计算的真菌群落之间的系统发育距离可以解释更多的方差,而不是非系统发育距离。这些系统发育指标也提高了我们区分微生物群落之间微小差异(效应量)的能力,尽管对于更大的效应量,结果与非系统发育方法相似。
这里提出的基于 Silva/UNITE 的 ghost tree 可以很容易地集成到现有的真菌分析管道中,以提高真菌群落差异的分辨率,并增强对建筑环境中这些群落的理解。ghost-tree 软件包还可以用于为其他提供不同分类分辨率的标记基因集开发系统发育树,或者用于将基因组树与扩增子树连接起来。
ghost-tree 可以通过 pip 进行安装。所有的源代码、文档和测试代码都可以在 BSD 许可证下在 https://github.com/JTFouquier/ghost-tree 获得。