Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Genet Epidemiol. 2011 Dec;35(8):790-9. doi: 10.1002/gepi.20628. Epub 2011 Sep 15.
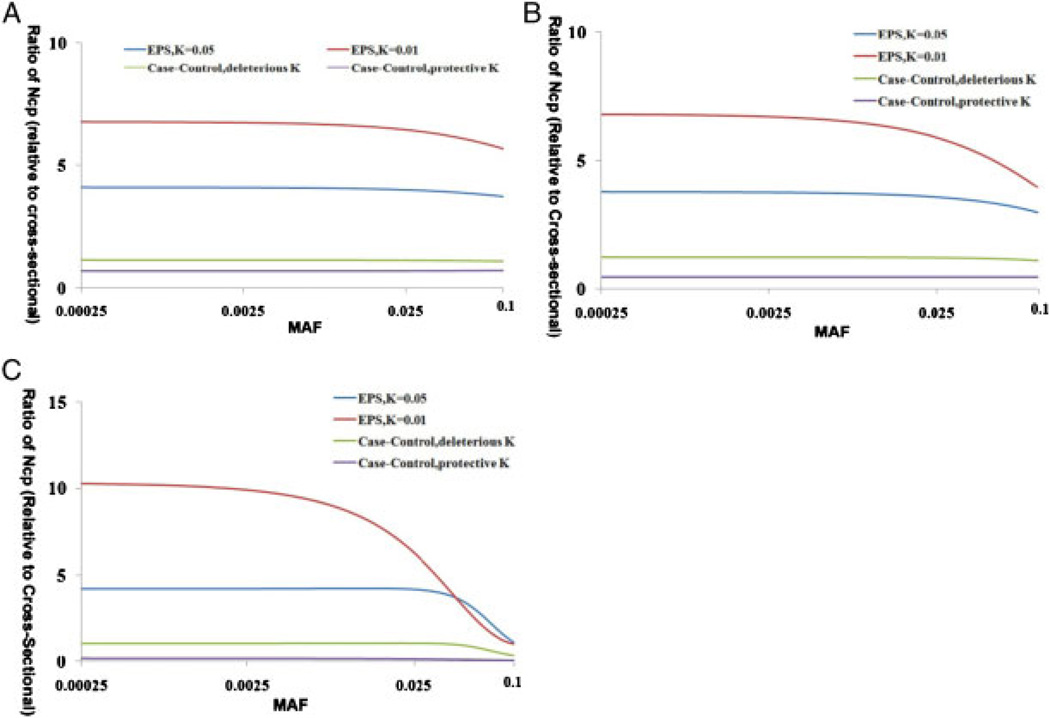
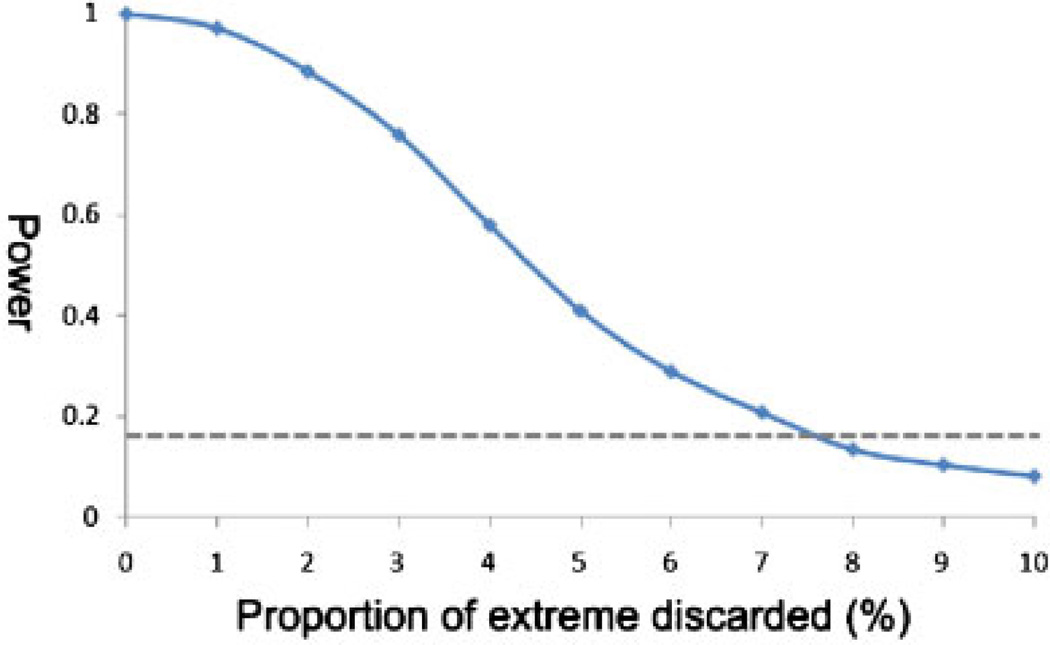
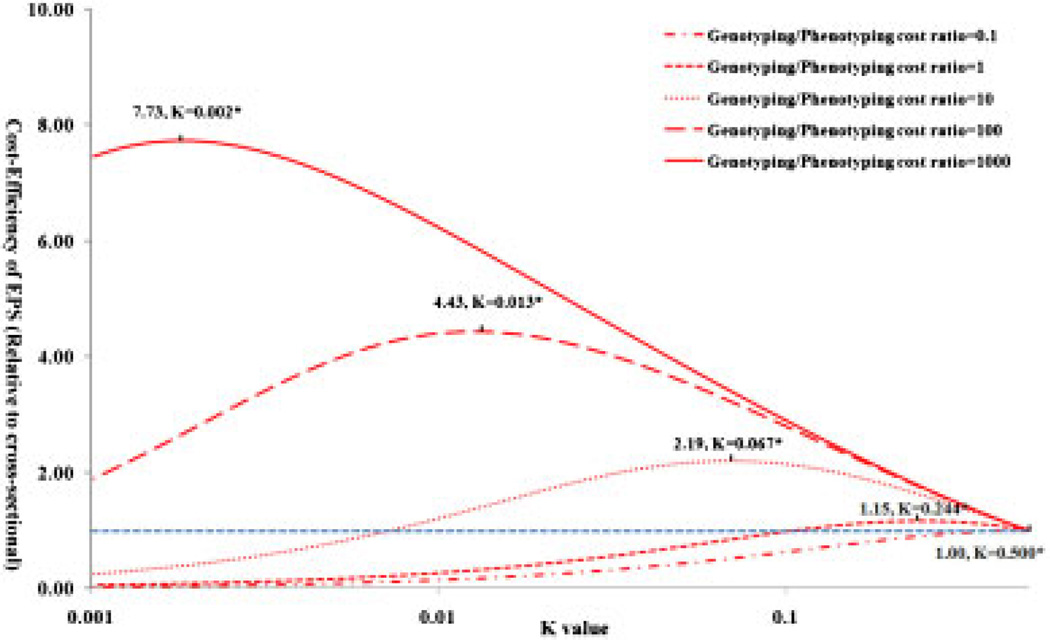
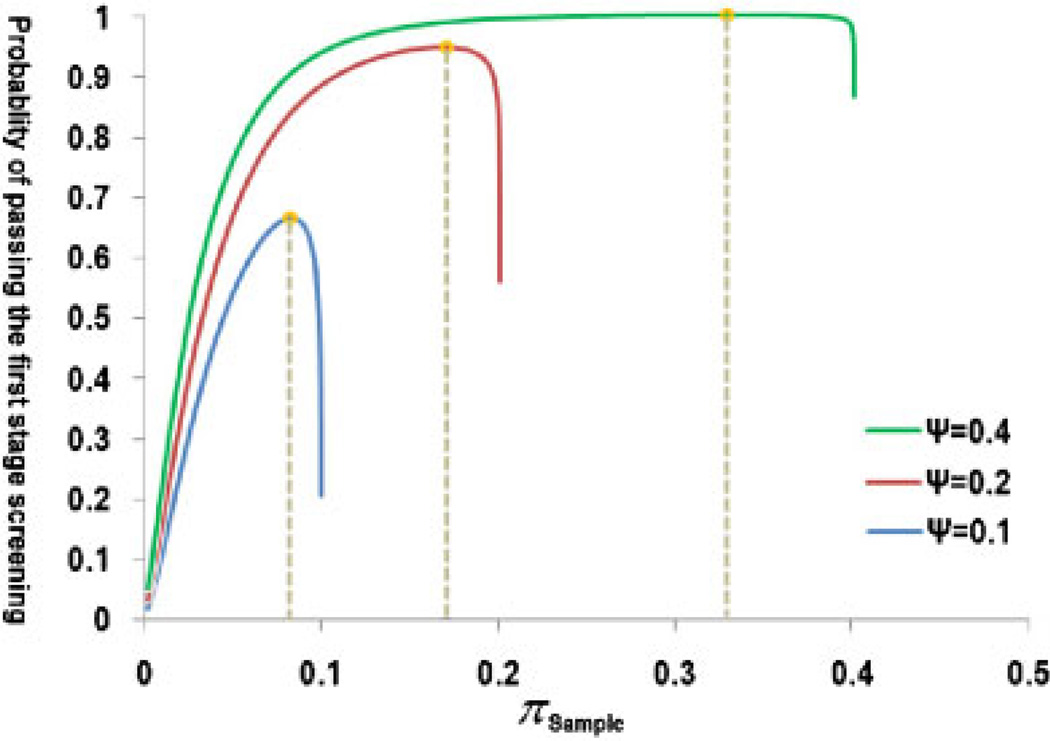
Variants identified in recent genome-wide association studies based on the common-disease common-variant hypothesis are far from fully explaining the hereditability of complex traits. Rare variants may, in part, explain some of the missing hereditability. Here, we explored the advantage of the extreme phenotype sampling in rare-variant analysis and refined this design framework for future large-scale association studies on quantitative traits. We first proposed a power calculation approach for a likelihood-based analysis method. We then used this approach to demonstrate the potential advantages of extreme phenotype sampling for rare variants. Next, we discussed how this design can influence future sequencing-based association studies from a cost-efficiency (with the phenotyping cost included) perspective. Moreover, we discussed the potential of a two-stage design with the extreme sample as the first stage and the remaining nonextreme subjects as the second stage. We demonstrated that this two-stage design is a cost-efficient alternative to the one-stage cross-sectional design or traditional two-stage design. We then discussed the analysis strategies for this extreme two-stage design and proposed a corresponding design optimization procedure. To address many practical concerns, for example measurement error or phenotypic heterogeneity at the very extremes, we examined an approach in which individuals with very extreme phenotypes are discarded. We demonstrated that even with a substantial proportion of these extreme individuals discarded, an extreme-based sampling can still be more efficient. Finally, we expanded the current analysis and design framework to accommodate the CMC approach where multiple rare-variants in the same gene region are analyzed jointly.
基于常见疾病常见变异假说的全基因组关联研究中鉴定的变异远远不能完全解释复杂性状的遗传性。罕见变异可能部分解释了一些缺失的遗传性。在这里,我们探讨了在罕见变异分析中极端表型抽样的优势,并对这种设计框架进行了改进,以用于未来对定量性状的大规模关联研究。我们首先提出了一种基于似然的分析方法的功效计算方法。然后,我们使用这种方法来证明极端表型抽样对罕见变异的潜在优势。接下来,我们从成本效益(包括表型成本)的角度讨论了这种设计如何影响未来基于测序的关联研究。此外,我们还讨论了将极端样本作为第一阶段,其余非极端样本作为第二阶段的两阶段设计的潜力。我们证明了这种两阶段设计是一种具有成本效益的替代方案,可替代单阶段横断面设计或传统的两阶段设计。然后,我们讨论了这种极端两阶段设计的分析策略,并提出了相应的设计优化程序。为了解决许多实际问题,例如在非常极端情况下的测量误差或表型异质性,我们研究了一种方法,即将具有非常极端表型的个体排除在外。我们证明,即使排除了相当一部分这些极端个体,基于极端的抽样仍然可以更有效。最后,我们扩展了当前的分析和设计框架,以适应 CMC 方法,其中可以联合分析同一基因区域中的多个罕见变异。