Kochańczyk Marek
Faculty of Physics, Jagiellonian University, ul, Reymonta 4, 30-059 Krakow, Poland.
BMC Struct Biol. 2011 Sep 18;11:34. doi: 10.1186/1472-6807-11-34.
Well-performing automated protein function recognition approaches usually comprise several complementary techniques. Beside constructing better consensus, their predictive power can be improved by either adding or refining independent modules that explore orthogonal features of proteins. In this work, we demonstrated how the exploration of global atomic distributions can be used to indicate functionally important residues.
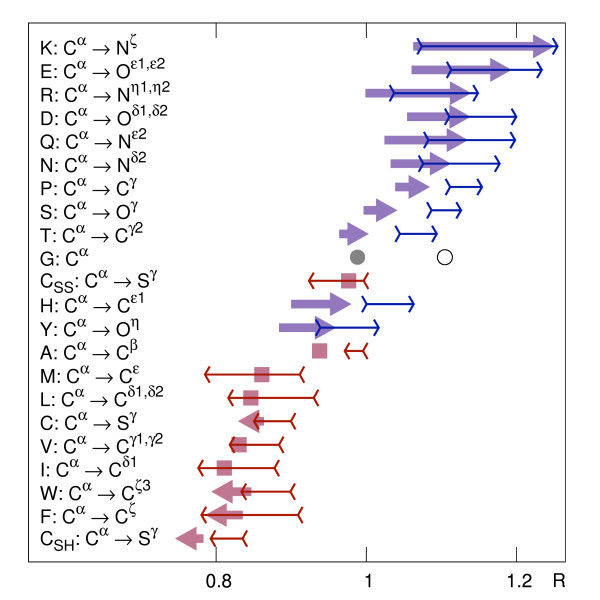
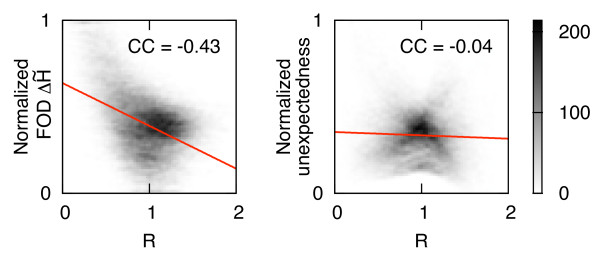
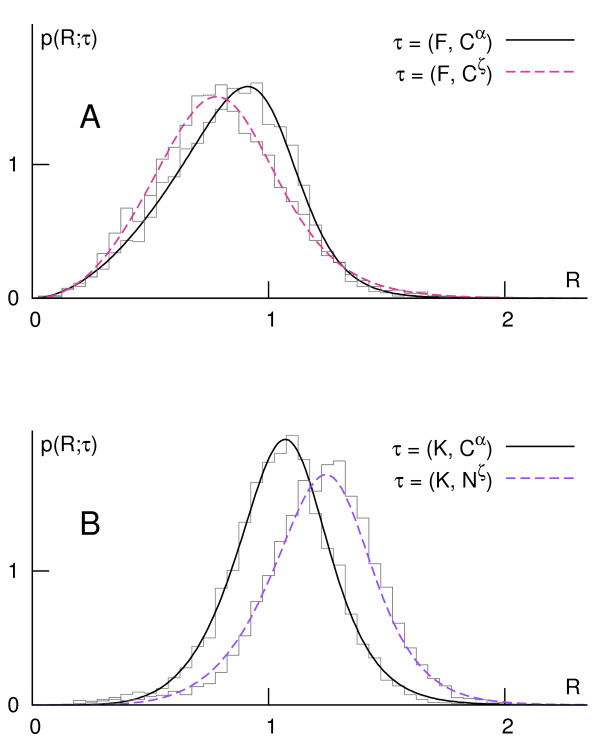
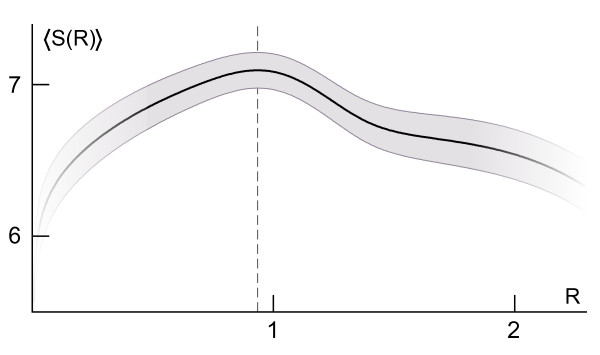
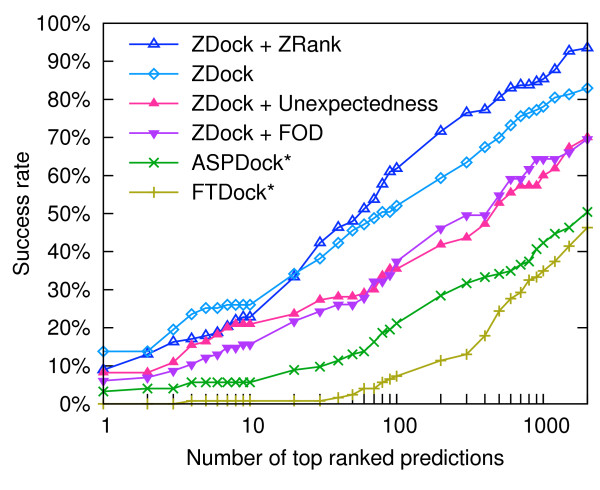
Using a set of carefully selected globular proteins, we parametrized continuous probability density functions describing preferred central distances of individual protein atoms. Relative preferred burials were estimated using mixture models of radial density functions dependent on the amino acid composition of a protein under consideration. The unexpectedness of extraordinary locations of atoms was evaluated in the information-theoretic manner and used directly for the identification of key amino acids. In the validation study, we tested capabilities of a tool built upon our approach, called SurpResi, by searching for binding sites interacting with ligands. The tool indicated multiple candidate sites achieving success rates comparable to several geometric methods. We also showed that the unexpectedness is a property of regions involved in protein-protein interactions, and thus can be used for the ranking of protein docking predictions. The computational approach implemented in this work is freely available via a Web interface at http://www.bioinformatics.org/surpresi.
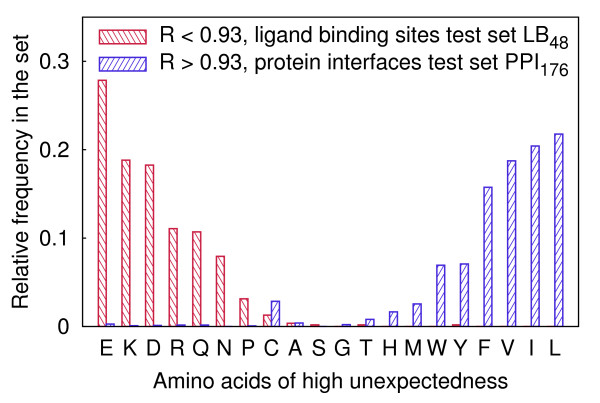
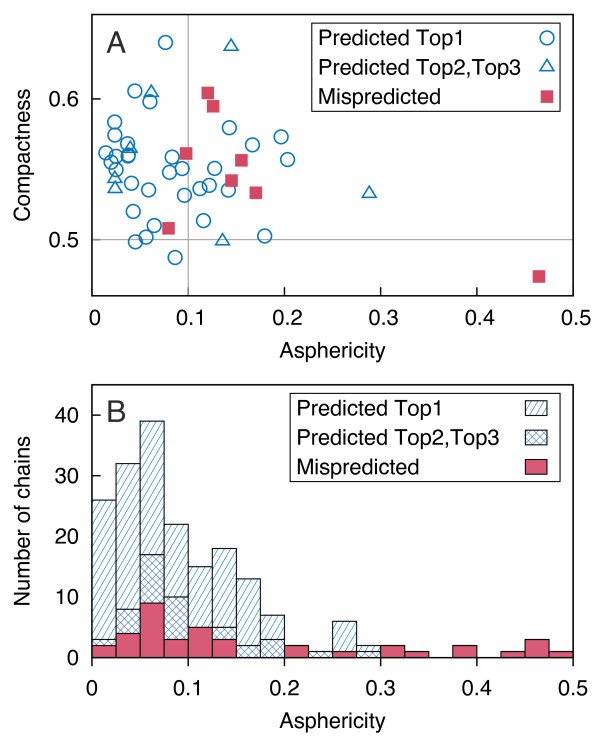
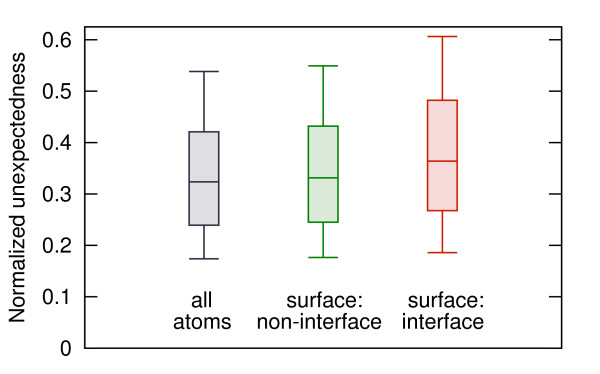
Probabilistic analysis of atomic central distances in globular proteins is capable of capturing distinct orientational preferences of amino acids as resulting from different sizes, charges and hydrophobic characters of their side chains. When idealized spatial preferences can be inferred from the sole amino acid composition of a protein, residues located in hydrophobically unfavorable environments can be easily detected. Such residues turn out to be often directly involved in binding ligands or interfacing with other proteins.
性能良好的自动化蛋白质功能识别方法通常由几种互补技术组成。除了构建更好的共识外,还可以通过添加或完善探索蛋白质正交特征的独立模块来提高其预测能力。在这项工作中,我们展示了如何利用对全局原子分布的探索来指示功能重要的残基。
我们使用一组精心挑选的球状蛋白质,对描述单个蛋白质原子优选中心距离的连续概率密度函数进行参数化。使用依赖于所考虑蛋白质氨基酸组成的径向密度函数混合模型来估计相对优选埋藏。以信息论的方式评估原子异常位置的意外性,并直接用于识别关键氨基酸。在验证研究中,我们通过搜索与配体相互作用的结合位点,测试了基于我们的方法构建的工具SurpResi的能力。该工具指出了多个候选位点,成功率与几种几何方法相当。我们还表明,意外性是参与蛋白质-蛋白质相互作用区域的一种属性,因此可用于对蛋白质对接预测进行排序。这项工作中实施的计算方法可通过网页界面(http://www.bioinformatics.org/surpresi)免费获得。
对球状蛋白质中原子中心距离的概率分析能够捕捉氨基酸由于其侧链不同大小、电荷和疏水特性而产生的独特取向偏好。当可以从蛋白质的单一氨基酸组成推断出理想的空间偏好时,位于疏水不利环境中的残基就很容易被检测到。事实证明,这些残基通常直接参与结合配体或与其他蛋白质相互作用。