Center for Microbial Genetics and Genomics, Northern Arizona University, Flagstaff, Arizona, USA.
PLoS Negl Trop Dis. 2011 Sep;5(9):e1319. doi: 10.1371/journal.pntd.0001319. Epub 2011 Sep 13.
Plague was introduced to Madagascar in 1898 and continues to be a significant human health problem. It exists mainly in the central highlands, but in the 1990s was reintroduced to the port city of Mahajanga, where it caused extensive human outbreaks. Despite its prevalence, the phylogeography and molecular epidemiology of Y. pestis in Madagascar has been difficult to study due to the great genetic similarity among isolates. We examine island-wide geographic-genetic patterns based upon whole-genome discovery of SNPs, SNP genotyping and hypervariable variable-number tandem repeat (VNTR) loci to gain insight into the maintenance and spread of Y. pestis in Madagascar.
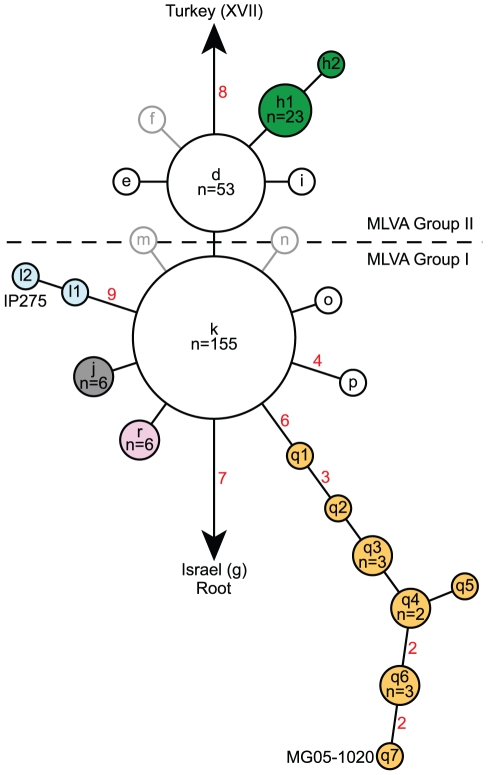
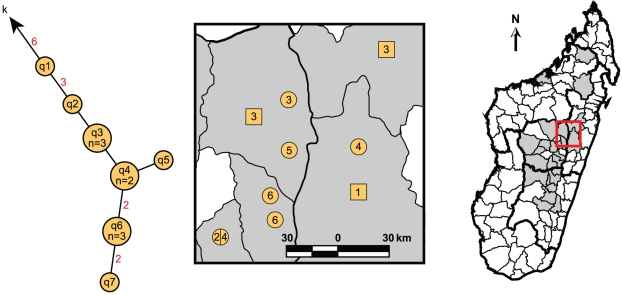
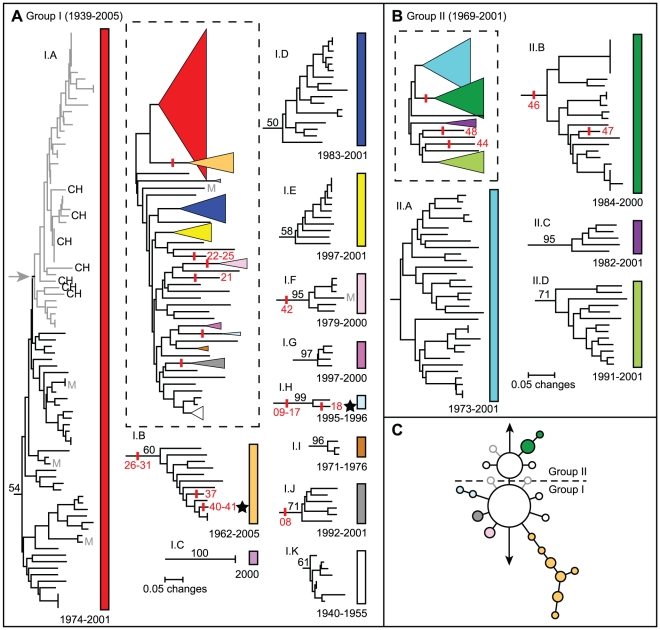
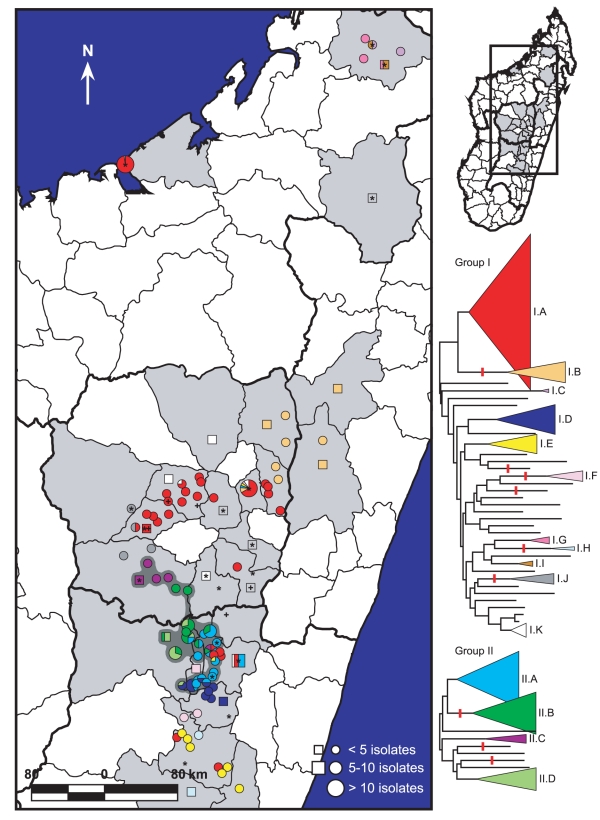
METHODOLOGY/PRINCIPAL FINDINGS: We analyzed a set of 262 Malagasy isolates using a set of 56 SNPs and a 43-locus multi-locus VNTR analysis (MLVA) system. We then analyzed the geographic distribution of the subclades and identified patterns related to the maintenance and spread of plague in Madagascar. We find relatively high levels of VNTR diversity in addition to several SNP differences. We identify two major groups, Groups I and II, which are subsequently divided into 11 and 4 subclades, respectively. Y. pestis appears to be maintained in several geographically separate subpopulations. There is also evidence for multiple long distance transfers of Y. pestis, likely human mediated. Such transfers have resulted in the reintroduction and establishment of plague in the port city of Mahajanga, where there is evidence for multiple transfers both from and to the central highlands.
CONCLUSIONS/SIGNIFICANCE: The maintenance and spread of Y. pestis in Madagascar is a dynamic and highly active process that relies on the natural cycle between the primary host, the black rat, and its flea vectors as well as human activity.
1898 年,鼠疫传入马达加斯加,至今仍是一个重大的人类健康问题。它主要存在于中部高原,但在 20 世纪 90 年代,它被重新引入港口城市马任加,在那里引发了广泛的人类疫情。尽管鼠疫在马达加斯加很普遍,但由于分离株之间存在很大的遗传相似性,其鼠疫耶尔森氏菌的系统地理学和分子流行病学一直难以研究。我们通过全基因组发现 SNP、SNP 基因分型和高变可变数串联重复(VNTR)位点,研究了整个岛屿的地理遗传模式,以深入了解鼠疫耶尔森氏菌在马达加斯加的维持和传播。
方法/主要发现:我们使用一组 56 个 SNP 和一个 43 个位点的多位点 VNTR 分析(MLVA)系统分析了一组 262 个马达加斯加分离株。然后,我们分析了亚分支的地理分布,并确定了与马达加斯加鼠疫维持和传播有关的模式。我们发现除了几个 SNP 差异外,VNTR 多样性也相当高。我们确定了两个主要的群组,I 组和 II 组,它们分别进一步分为 11 个和 4 个亚分支。鼠疫耶尔森氏菌似乎在几个地理上分开的亚种群中得到维持。也有证据表明鼠疫耶尔森氏菌发生了多次长距离转移,可能是人为介导的。这种转移导致鼠疫在港口城市马任加的重新引入和建立,在那里有证据表明从中部高原既有输入也有输出。
结论/意义:鼠疫耶尔森氏菌在马达加斯加的维持和传播是一个动态的、高度活跃的过程,它依赖于主要宿主黑鼠及其跳蚤媒介之间的自然循环以及人类活动。