Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology and University of Tuebingen, Stuttgart, Germany.
PLoS One. 2011;6(9):e25139. doi: 10.1371/journal.pone.0025139. Epub 2011 Sep 20.
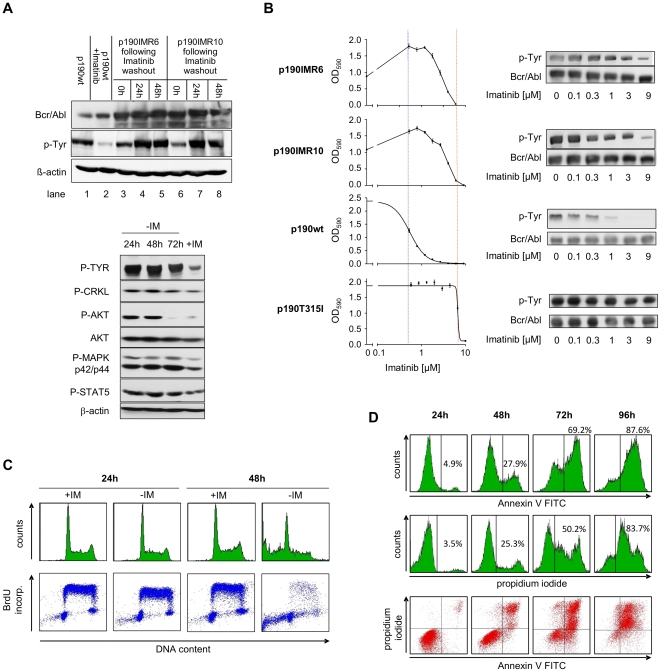
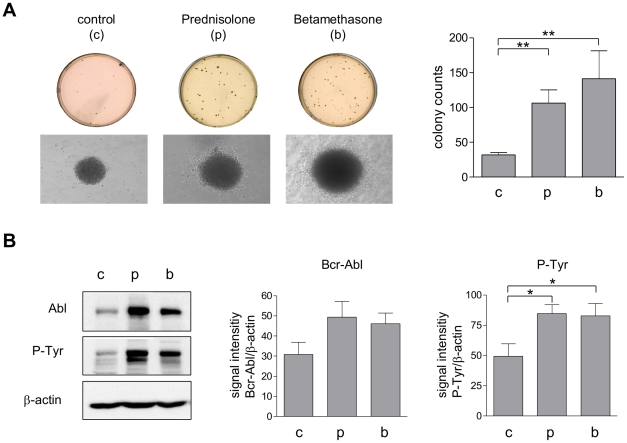
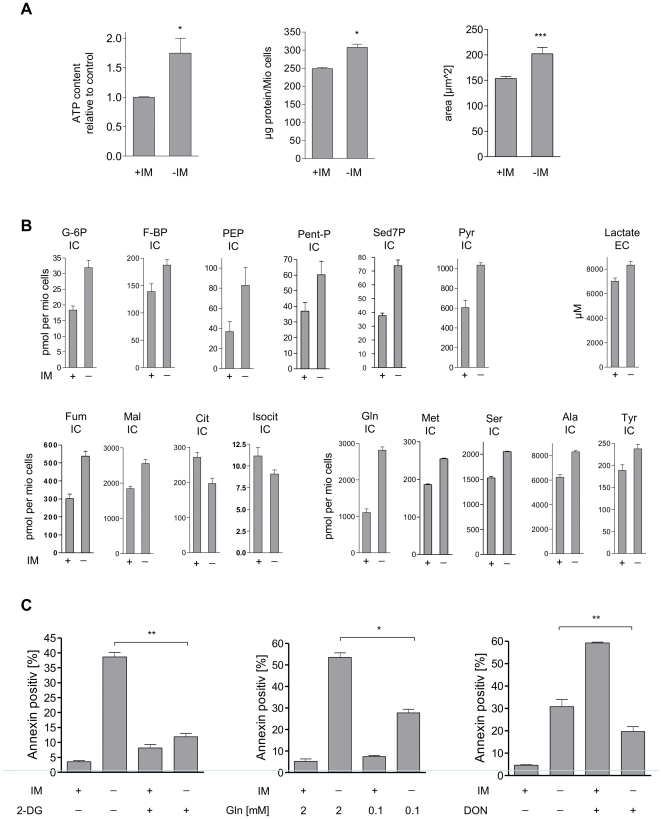
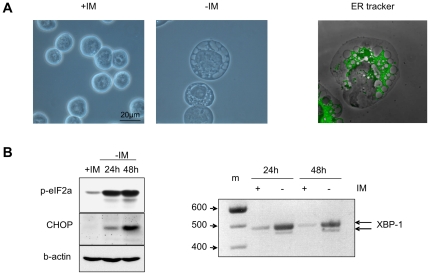
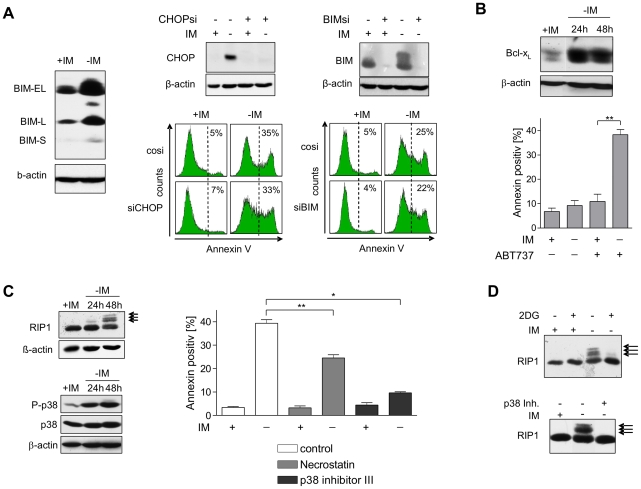
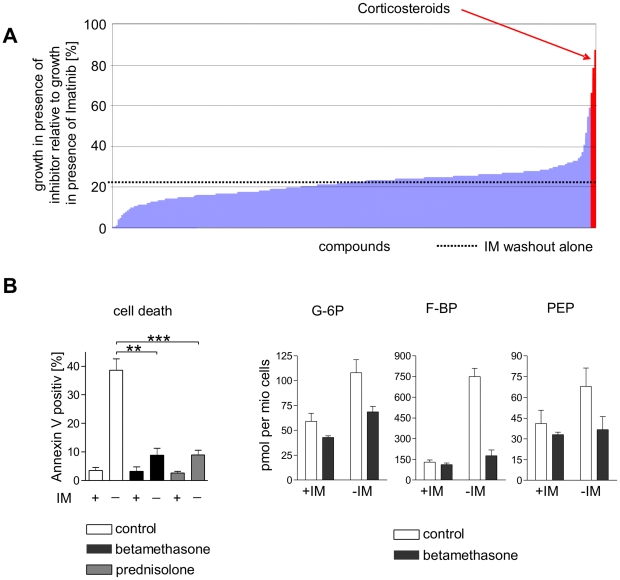
In response to deregulated oncogene activation, mammalian cells activate disposal programs such as programmed cell death. To investigate the mechanisms behind this oncogenic stress response we used Bcr-Abl over-expressing cells cultivated in presence of imatinib. Imatinib deprivation led to rapid induction of Bcr-Abl activity and over-stimulation of PI3K/Akt-, Ras/MAPK-, and JAK/STAT pathways. This resulted in a delayed necrosis-like cell death starting not before 48 hours after imatinib withdrawal. Cell death was preceded by enhanced glycolysis, glutaminolysis, and amino acid metabolism leading to elevated ATP and protein levels. This enhanced metabolism could be linked to induction of cell death as inhibition of glycolysis or glutaminolysis was sufficient to sustain cell viability. Therefore, these data provide first evidence that metabolic changes induced by Bcr-Abl hyper-activation are important mediators of oncogenic stress-induced cell death.During the first 30 hours after imatinib deprivation, Bcr-Abl hyper-activation did not affect proliferation but resulted in cellular swelling, vacuolization, and induction of eIF2α phosphorylation, CHOP expression, as well as alternative splicing of XPB, indicating endoplasmic reticulum stress response. Cell death was dependent on p38 and RIP1 signaling, whereas classical death effectors of ER stress, namely CHOP-BIM were antagonized by concomitant up-regulation of Bcl-xL.Screening of 1,120 compounds for their potential effects on oncogenic stress-induced cell death uncovered that corticosteroids antagonize cell death upon Bcr-Abl hyper-activation by normalizing cellular metabolism. This protective effect is further demonstrated by the finding that corticosteroids rendered lymphocytes permissive to the transforming activity of Bcr-Abl. As corticosteroids are used together with imatinib for treatment of Bcr-Abl positive acute lymphoblastic leukemia these data could have important implications for the design of combination therapy protocols.In conclusion, excessive induction of Warburg type metabolic alterations can cause cell death. Our data indicate that these metabolic changes are major mediators of oncogenic stress induced by Bcr-Abl.
针对失调的致癌基因激活,哺乳动物细胞会激活程序性细胞死亡等处置程序。为了研究这种致癌应激反应背后的机制,我们使用了在伊马替尼存在的情况下过度表达 Bcr-Abl 的细胞进行培养。伊马替尼剥夺导致 Bcr-Abl 活性的快速诱导和 PI3K/Akt、Ras/MAPK 和 JAK/STAT 途径的过度刺激。这导致了类似于迟发性坏死的细胞死亡,在伊马替尼撤出后 48 小时之前不会开始。细胞死亡之前是增强的糖酵解、谷氨酰胺分解和氨基酸代谢,导致 ATP 和蛋白质水平升高。这种增强的代谢可能与细胞死亡的诱导有关,因为抑制糖酵解或谷氨酰胺分解足以维持细胞活力。因此,这些数据首次提供了证据,证明由 Bcr-Abl 过度激活引起的代谢变化是致癌应激诱导细胞死亡的重要介质。在伊马替尼剥夺后的头 30 小时内,Bcr-Abl 的过度激活不会影响增殖,但会导致细胞肿胀、空泡化和 eIF2α 磷酸化、CHOP 表达以及 XPB 的选择性剪接的诱导,表明内质网应激反应。细胞死亡依赖于 p38 和 RIP1 信号,而内质网应激的经典死亡效应物,即 CHOP-BIM,则被 Bcl-xL 的同时上调拮抗。对 1120 种化合物进行筛选,以研究它们对致癌应激诱导的细胞死亡的潜在影响,发现皮质类固醇通过使细胞代谢正常化来拮抗 Bcr-Abl 过度激活时的细胞死亡。通过发现皮质类固醇使淋巴细胞对 Bcr-Abl 的转化活性变得易感性,进一步证明了这种保护作用。由于皮质类固醇与伊马替尼一起用于治疗 Bcr-Abl 阳性急性淋巴细胞白血病,这些数据可能对联合治疗方案的设计具有重要意义。总之,过度诱导沃伯格型代谢改变会导致细胞死亡。我们的数据表明,这些代谢变化是 Bcr-Abl 诱导的致癌应激的主要介质。