Immunology Group, International Centre for Genetic Engineering and Biotechnology, Aruna Asaf Ali Marg, New Delhi 110067, India.
J Biol Chem. 2011 Nov 18;286(46):40307-19. doi: 10.1074/jbc.M111.266239. Epub 2011 Sep 27.
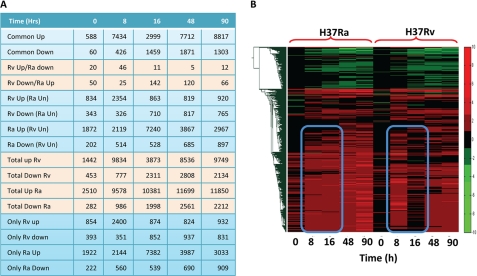
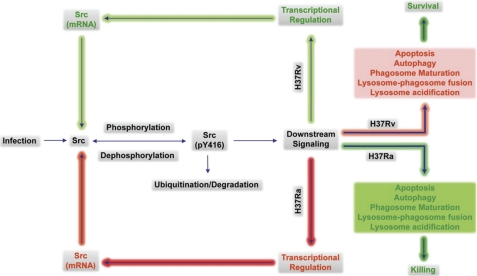
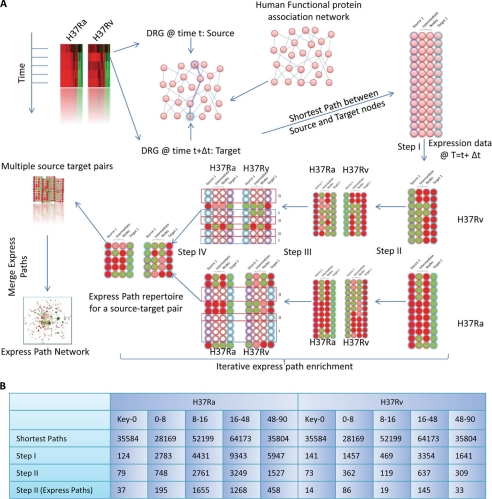
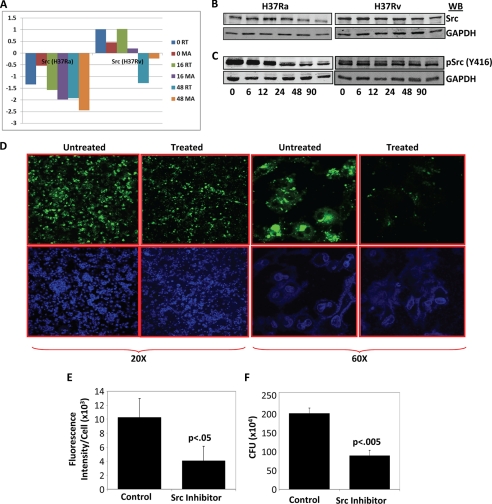
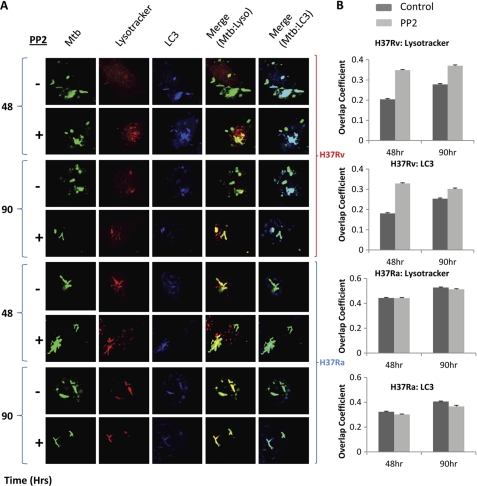
Global gene expression profiling has emerged as a major tool in understanding complex response patterns of biological systems to perturbations. However, a lack of unbiased analytical approaches has restricted the utility of complex microarray data to gain novel system level insights. Here we report a strategy, express path analysis (EPA), that helps to establish various pathways differentially recruited to achieve specific cellular responses under contrasting environmental conditions in an unbiased manner. The analysis superimposes differentially regulated genes between contrasting environments onto the network of functional protein associations followed by a series of iterative enrichments and network analysis. To test the utility of the approach, we infected THP1 macrophage cells with a virulent Mycobacterium tuberculosis strain (H37Rv) or the attenuated non-virulent strain H37Ra as contrasting perturbations and generated the temporal global expression profiles. EPA of the results provided details of response-specific and time-dependent host molecular network perturbations. Further analysis identified tyrosine kinase Src as the major regulatory hub discriminating the responses between wild-type and attenuated Mtb infection. We were then able to verify this novel role of Src experimentally and show that Src executes its role through regulating two vital antimicrobial processes of the host cells (i.e. autophagy and acidification of phagolysosome). These results bear significant potential for developing novel anti-tuberculosis therapy. We propose that EPA could prove extremely useful in understanding complex cellular responses for a variety of perturbations, including pathogenic infections.
全球基因表达谱分析已成为理解生物系统对干扰的复杂反应模式的主要工具。然而,缺乏无偏分析方法限制了复杂微阵列数据在获得新的系统水平见解方面的应用。在这里,我们报告了一种策略,表达路径分析(EPA),该策略有助于以无偏的方式建立各种途径,以实现特定的细胞反应,从而获得各种途径。该分析将对比环境下差异调节的基因叠加到功能蛋白关联网络上,然后进行一系列迭代富集和网络分析。为了测试该方法的实用性,我们用一种毒力结核分枝杆菌(H37Rv)或减毒非毒力株 H37Ra 感染 THP1 巨噬细胞作为对比干扰,并生成了时间上的全局表达谱。EPA 对结果的分析提供了宿主分子网络对特定反应和时间依赖性干扰的详细信息。进一步的分析确定酪氨酸激酶Src 是区分野生型和减毒 Mtb 感染反应的主要调节枢纽。然后,我们能够通过实验验证 Src 的这一新颖作用,并表明 Src 通过调节宿主细胞的两个重要抗菌过程(即自噬和吞噬溶酶体酸化)来执行其作用。这些结果为开发新型抗结核疗法提供了重要的潜力。我们提出,EPA 可以证明在理解各种干扰(包括致病性感染)的复杂细胞反应方面非常有用。