Department of Cell Biology, Harvard Medical School, Boston, Massachusetts, USA.
Nat Methods. 2011 Oct 2;8(11):937-40. doi: 10.1038/nmeth.1714.
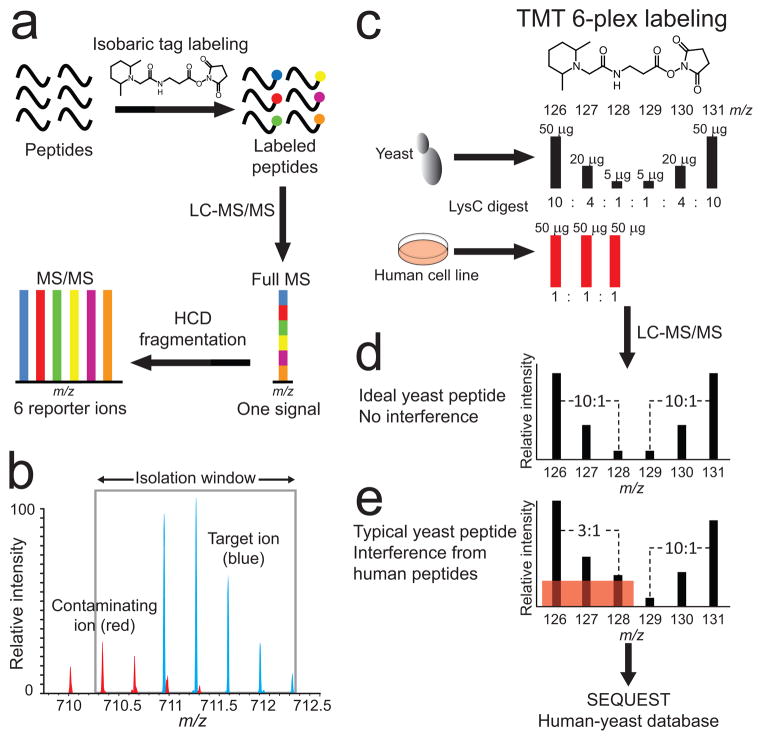
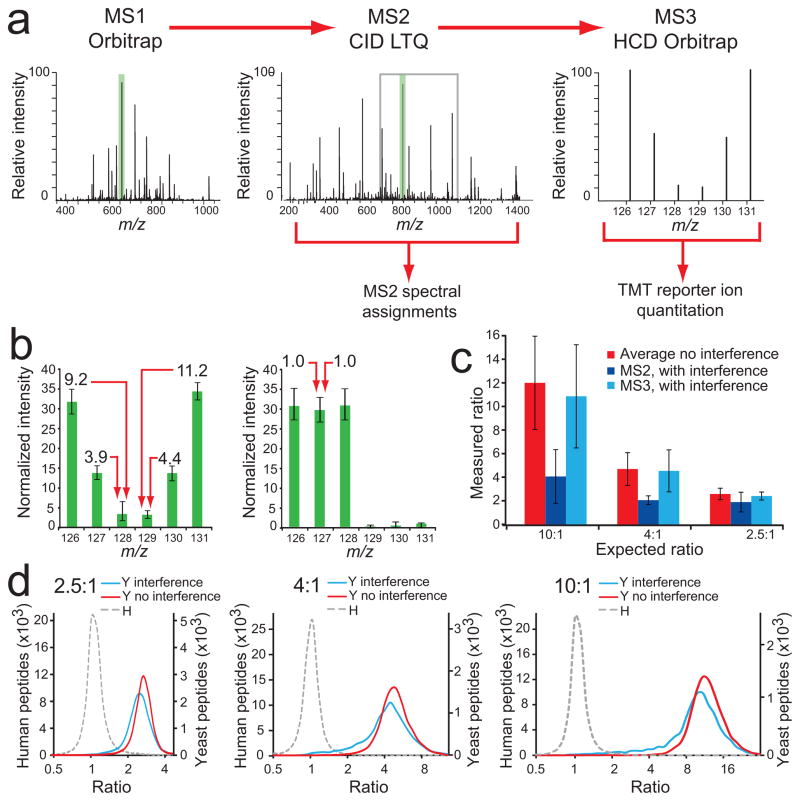
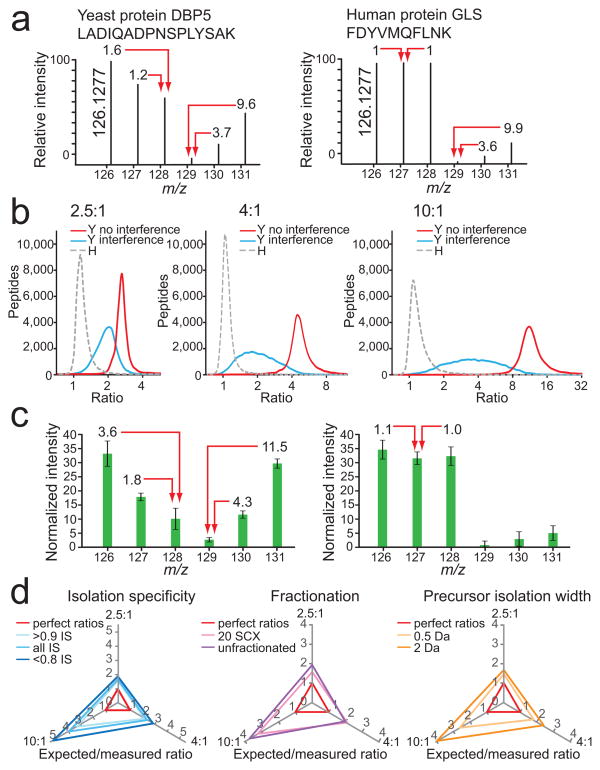
Quantitative mass spectrometry-based proteomics is highly versatile but not easily multiplexed. Isobaric labeling strategies allow mass spectrometry-based multiplexed proteome quantification; however, ratio distortion owing to protein quantification interference is a common effect. We present a two-proteome model (mixture of human and yeast proteins) in a sixplex isobaric labeling system to fully document the interference effect, and we report that applying triple-stage mass spectrometry (MS3) almost completely eliminates interference.
基于定量质谱的蛋白质组学具有高度的通用性,但不容易实现多重化。同重标记策略允许基于质谱的多重蛋白质组定量;然而,由于蛋白质定量干扰导致的比率失真是一种常见的影响。我们提出了一个在六重同重标记系统中的双蛋白质组模型(人蛋白和酵母蛋白的混合物),以充分记录干扰效应,并报告说应用三级质谱(MS3)几乎可以完全消除干扰。