Department of Medicine, Stanford University Medical Center, Palo Alto, CA, USA.
Nucleic Acids Res. 2011 Dec;39(22):e151. doi: 10.1093/nar/gkr773. Epub 2011 Sep 30.
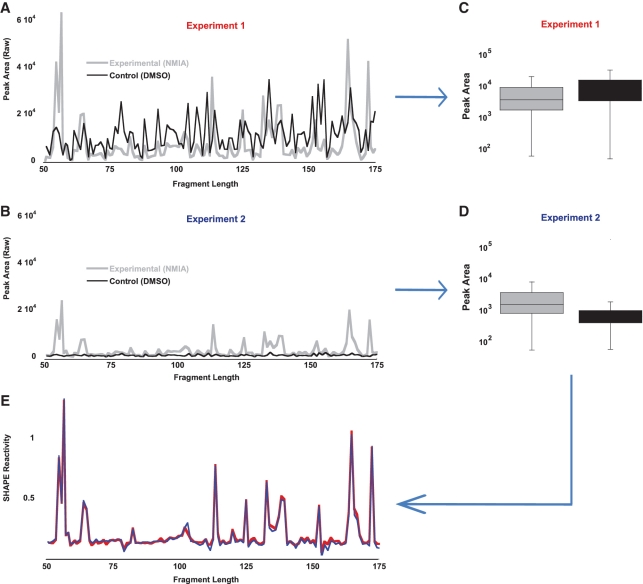
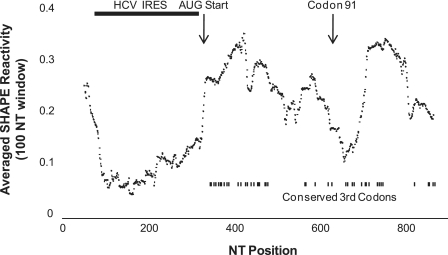
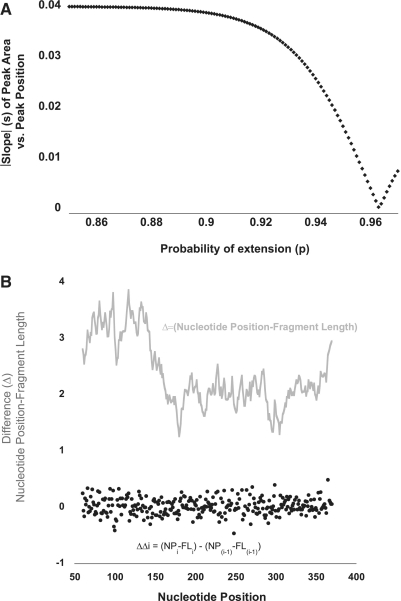
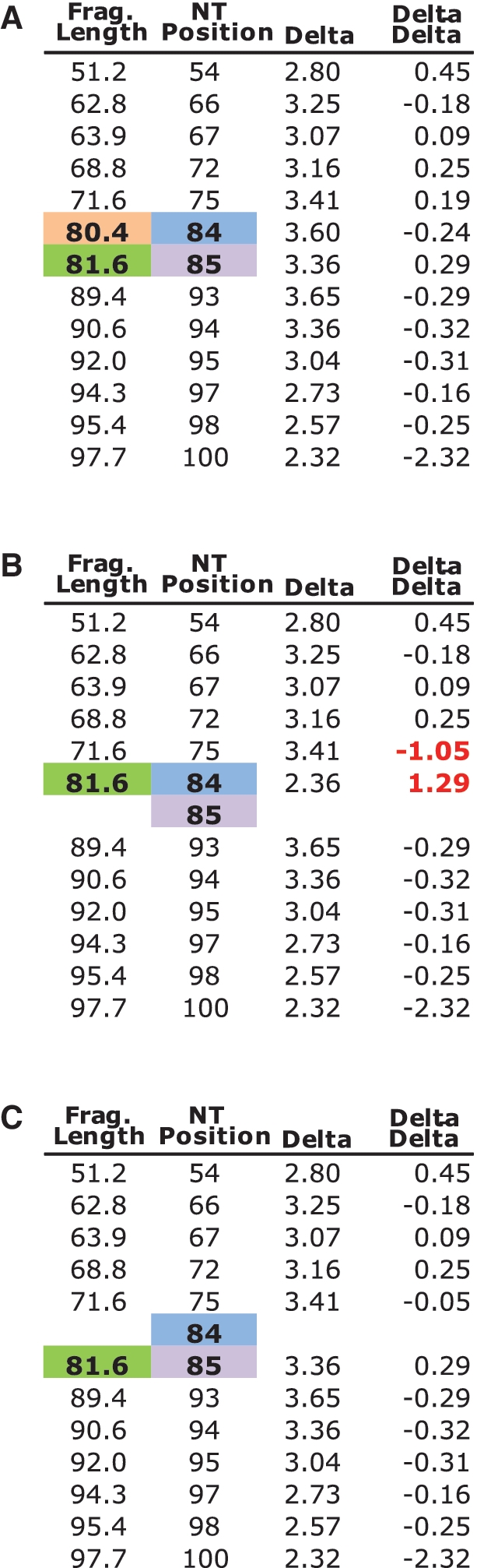
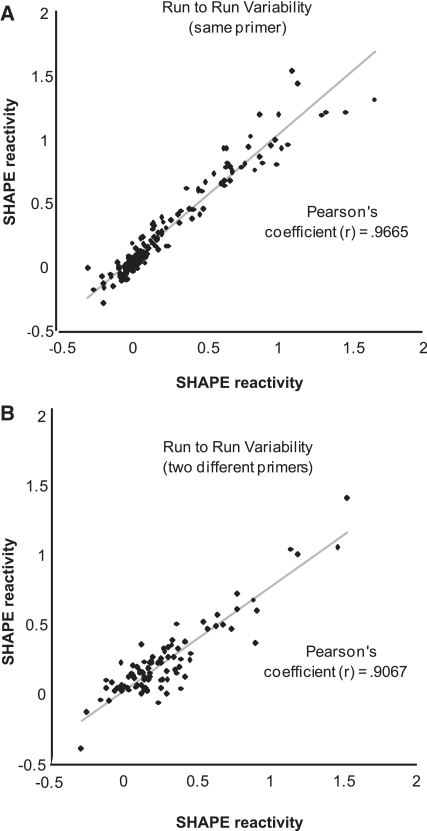
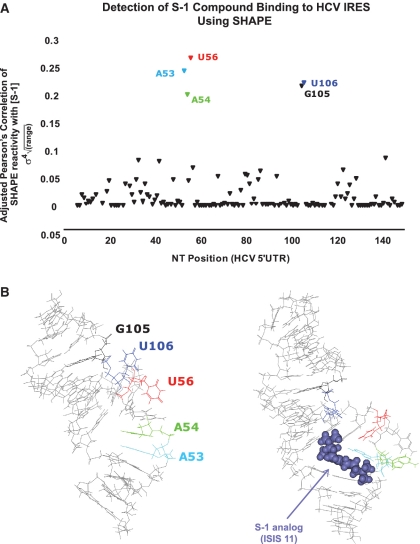
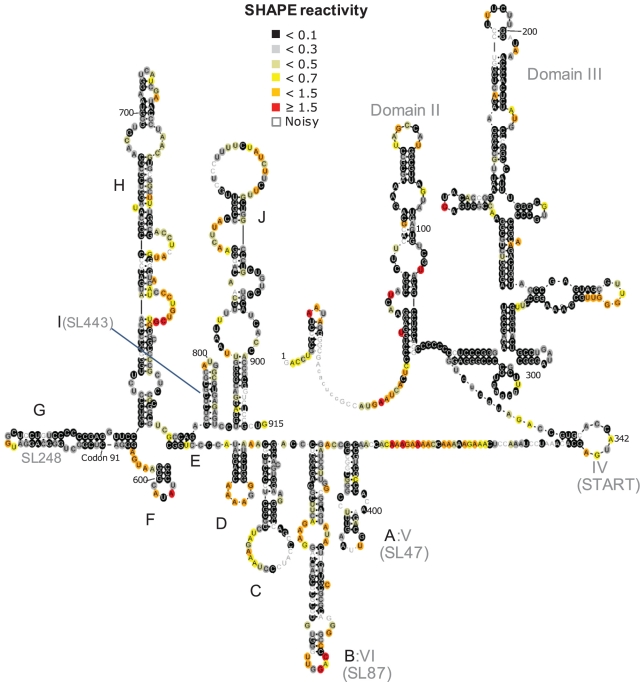
SHAPE (Selective 2'-hydroxyl acylation analysed by primer extension) technology has emerged as one of the leading methods of determining RNA secondary structure at the nucleotide level. A significant bottleneck in using SHAPE is the complex and time-consuming data processing that is required. We present here a modified data collection method and a series of algorithms, embodied in a program entitled Fast Analysis of SHAPE traces (FAST), which significantly reduces processing time. We have used this method to resolve the secondary structure of the first ~900 nt of the hepatitis C virus (HCV) genome, including the entire core gene. We have also demonstrated the ability of SHAPE/FAST to detect the binding of a small molecule inhibitor to the HCV internal ribosomal entry site (IRES). In conclusion, FAST allows for high-throughput data processing to match the current high-throughput generation of data possible with SHAPE, reducing the barrier to determining the structure of RNAs of interest.
选择性 2'-羟基酰化分析引物延伸(SHAPE)技术已成为在核苷酸水平上确定 RNA 二级结构的主要方法之一。在使用 SHAPE 时,一个显著的瓶颈是需要复杂且耗时的数据处理。我们在这里提出了一种改良的数据收集方法和一系列算法,这些算法体现在一个名为快速分析 SHAPE 轨迹(FAST)的程序中,该程序显著缩短了处理时间。我们已经使用该方法解析了丙型肝炎病毒(HCV)基因组前约 900 个核苷酸的二级结构,包括整个核心基因。我们还证明了 SHAPE/FAST 检测小分子抑制剂与 HCV 内部核糖体进入位点(IRES)结合的能力。总之,FAST 允许进行高通量数据处理,以匹配当前可能通过 SHAPE 生成的高通量数据,从而降低确定感兴趣 RNA 结构的障碍。