Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, Michigan 48824, USA.
Toxicol Sci. 2012 Jan;125(1):126-33. doi: 10.1093/toxsci/kfr266. Epub 2011 Oct 9.
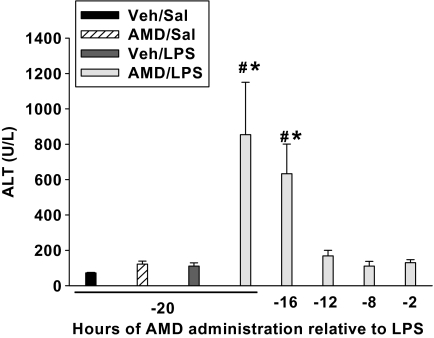
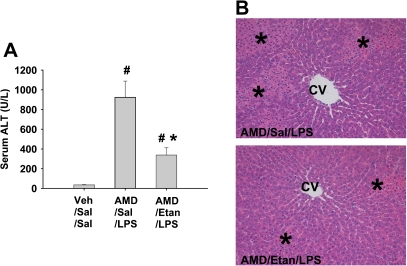
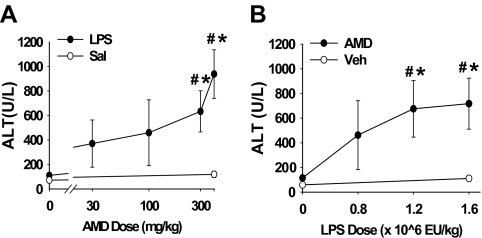
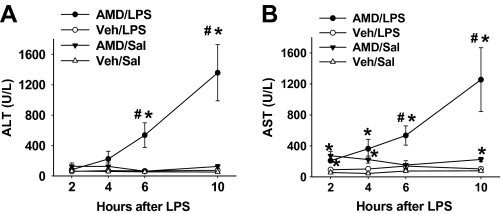
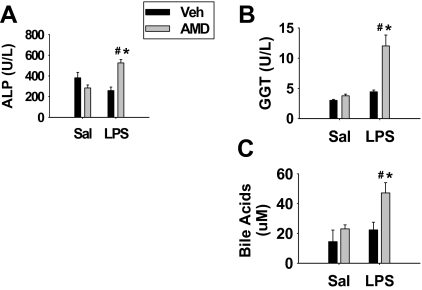
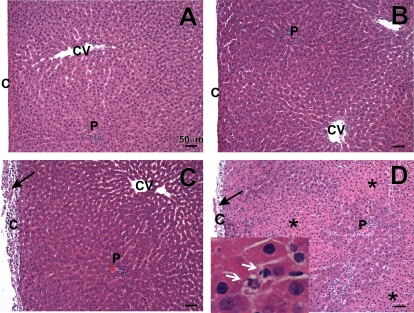
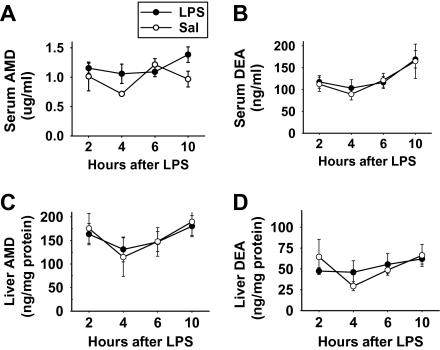
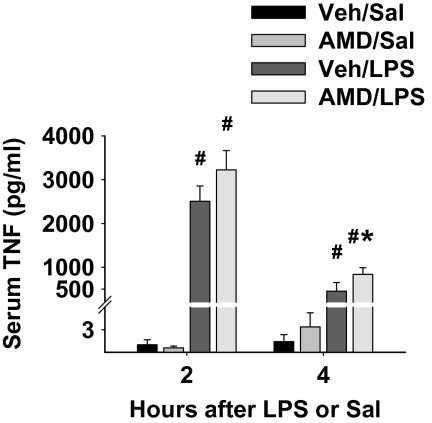
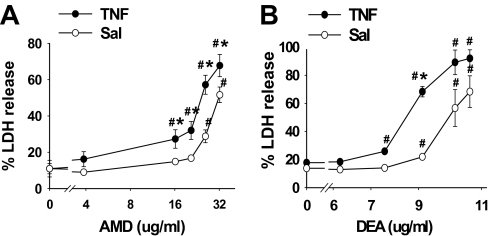
Amiodarone [2-butyl-3-(3',5'-diiodo-4'α-diethylaminoethoxybenzoyl)-benzofuran] (AMD), a class III antiarrhythmic drug, is known to cause idiosyncratic hepatotoxic reactions in human patients. One hypothesis for the etiology of idiosyncratic adverse drug reactions is that a concurrent inflammatory stress results in decreased threshold for drug toxicity. To explore this hypothesis in an animal model, male Sprague-Dawley rats were treated with nonhepatotoxic doses of AMD or its vehicle and with saline vehicle or lipopolysaccharide (LPS) to induce low-level inflammation. Elevated alanine aminotransferase (ALT), aspartate aminotransferase, alkaline phosphatase, and gamma-glutamyltransferase activities as well as increased total bile acid concentrations in serum and midzonal hepatocellular necrosis were observed only in AMD/LPS-cotreated rats. The time interval between AMD and LPS administration was critical: AMD injected 16 h before LPS led to liver injury, whereas AMD injected 2-12 h before LPS failed to cause this response. The increase in ALT activity in AMD/LPS cotreatment showed a clear dose-response relationship with AMD as well as LPS. The metabolism and hepatic accumulation of AMD were not affected by LPS coexposure. Serum concentration of tumor necrosis factor-alpha (TNF) was significantly increased by LPS and was slightly prolonged by AMD. In Hepac1c7 cells, addition of TNF potentiated the cytotoxicity of both AMD and its primary metabolite, mono-N-desethylamiodarone. In vivo inhibition of TNF signaling by etanercept attenuated the AMD/LPS-induced liver injury in rats. In summary, AMD treatment during modest inflammation induced severe hepatotoxicity in rats, and TNF contributed to the induction of liver injury in this animal model of idiosyncratic AMD-induced liver injury.
胺碘酮[2-丁基-3-(3',5'-二碘-4'α-二乙氨基乙氧基苯甲酰基)苯并呋喃](AMD)是一种 III 类抗心律失常药物,已知会导致人类患者出现特发性肝毒性反应。特发性药物不良反应病因的一个假设是,同时发生的炎症应激会降低药物毒性的阈值。为了在动物模型中探索这一假设,雄性 Sprague-Dawley 大鼠接受非肝毒性剂量的 AMD 或其载体以及生理盐水载体或脂多糖(LPS)处理,以诱导低水平炎症。仅在 AMD/LPS 共同处理的大鼠中观察到血清中天冬氨酸转氨酶(ALT)、丙氨酸转氨酶、碱性磷酸酶和γ-谷氨酰转移酶活性升高以及总胆汁酸浓度升高和中带肝细胞坏死。AMD 和 LPS 给药之间的时间间隔是关键的:AMD 在 LPS 前 16 小时注射会导致肝损伤,而 AMD 在 LPS 前 2-12 小时注射则不会引起这种反应。在 AMD/LPS 共同处理中,ALT 活性的增加与 AMD 以及 LPS 呈明显的剂量反应关系。LPS 共暴露并未影响 AMD 的代谢和肝蓄积。LPS 显著增加血清肿瘤坏死因子-α(TNF)浓度,并使 AMD 略有延长。在 Hepac1c7 细胞中,TNF 的添加增强了 AMD 及其主要代谢物单-N-去乙基胺碘酮的细胞毒性。体内 TNF 信号通路的抑制通过依那西普减弱了大鼠 AMD/LPS 诱导的肝损伤。总之,在适度炎症期间给予 AMD 治疗会导致大鼠严重肝毒性,而 TNF 有助于该动物模型中特发性 AMD 诱导的肝损伤的诱导。