Department of Computer Science, Georgia State University, Atlanta, GA 30303, USA.
BMC Bioinformatics. 2011;12 Suppl 6(Suppl 6):S1. doi: 10.1186/1471-2105-12-S6-S1. Epub 2011 Jul 28.
RNA viruses infecting a host usually exist as a set of closely related sequences, referred to as quasispecies. The genomic diversity of viral quasispecies is a subject of great interest, particularly for chronic infections, since it can lead to resistance to existing therapies. High-throughput sequencing is a promising approach to characterizing viral diversity, but unfortunately standard assembly software was originally designed for single genome assembly and cannot be used to simultaneously assemble and estimate the abundance of multiple closely related quasispecies sequences.
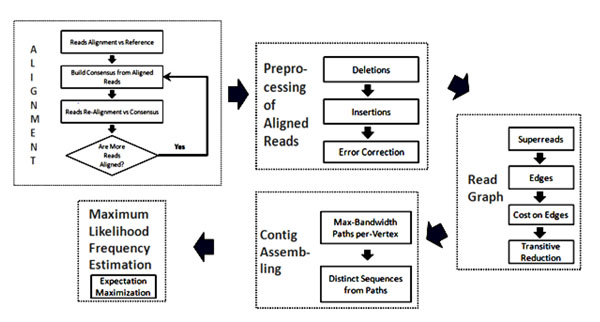
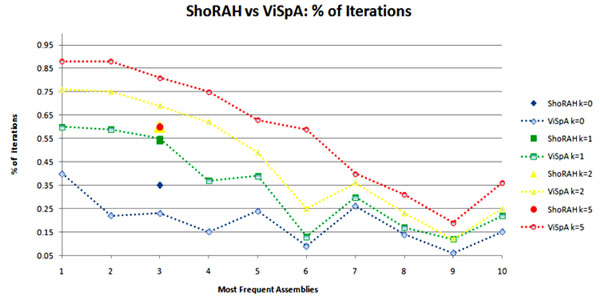
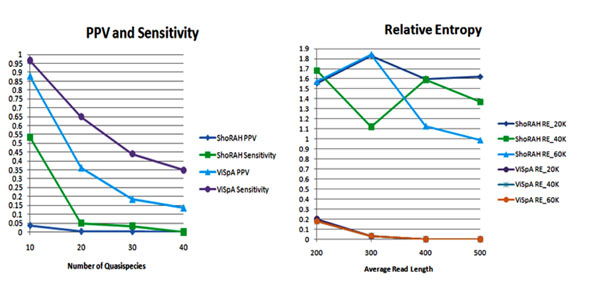
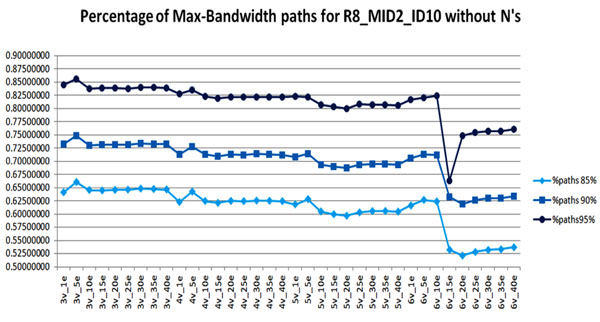
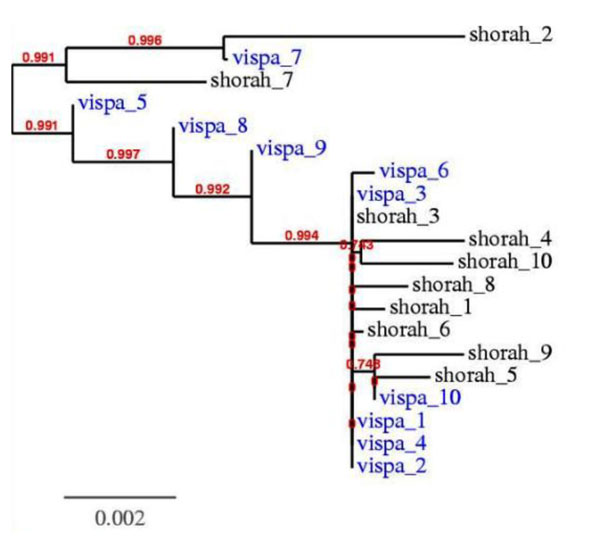
In this paper, we introduce a new Viral Spectrum Assembler (ViSpA) method for quasispecies spectrum reconstruction and compare it with the state-of-the-art ShoRAH tool on both simulated and real 454 pyrosequencing shotgun reads from HCV and HIV quasispecies. Experimental results show that ViSpA outperforms ShoRAH on simulated error-free reads, correctly assembling 10 out of 10 quasispecies and 29 sequences out of 40 quasispecies. While ShoRAH has a significant advantage over ViSpA on reads simulated with sequencing errors due to its advanced error correction algorithm, ViSpA is better at assembling the simulated reads after they have been corrected by ShoRAH. ViSpA also outperforms ShoRAH on real 454 reads. Indeed, 7 most frequent sequences reconstructed by ViSpA from a real HCV dataset are viable (do not contain internal stop codons), and the most frequent sequence was within 1% of the actual open reading frame obtained by cloning and Sanger sequencing. In contrast, only one of the sequences reconstructed by ShoRAH is viable. On a real HIV dataset, ShoRAH correctly inferred only 2 quasispecies sequences with at most 4 mismatches whereas ViSpA correctly reconstructed 5 quasispecies with at most 2 mismatches, and 2 out of 5 sequences were inferred without any mismatches. ViSpA source code is available at http://alla.cs.gsu.edu/~software/VISPA/vispa.html.
ViSpA enables accurate viral quasispecies spectrum reconstruction from 454 pyrosequencing reads. We are currently exploring extensions applicable to the analysis of high-throughput sequencing data from bacterial metagenomic samples and ecological samples of eukaryote populations.
感染宿主的 RNA 病毒通常存在一组密切相关的序列,称为准种。病毒准种的基因组多样性是一个非常感兴趣的课题,特别是对于慢性感染,因为它可能导致对现有治疗方法的耐药性。高通量测序是一种很有前途的方法来描述病毒多样性,但不幸的是,标准的组装软件最初是为单基因组组装设计的,不能同时组装和估计多个密切相关的准种序列的丰度。
本文介绍了一种新的病毒频谱组装(ViSpA)方法,用于准种谱重建,并将其与最先进的 ShoRAH 工具在 HCV 和 HIV 准种的模拟和真实 454 焦磷酸测序 shotgun 读取上进行了比较。实验结果表明,在无错误模拟读取中,ViSpA 比 ShoRAH 表现更好,正确组装了 10 个准种中的 10 个,以及 40 个准种中的 29 个序列。虽然 ShoRAH 由于其先进的纠错算法,在具有测序错误的模拟读取上优于 ViSpA,但在经过 ShoRAH 纠正后的模拟读取上,ViSpA 则更好。ViSpA 在真实的 454 读取上也优于 ShoRAH。事实上,从真实的 HCV 数据集重建的 ViSpA 中 7 个最常见的序列是可行的(不包含内部终止密码子),并且最常见的序列与通过克隆和 Sanger 测序获得的实际开放阅读框相差 1%以内。相比之下,只有 ShoRAH 重建的一个序列是可行的。在真实的 HIV 数据集上,ShoRAH 仅正确推断了最多有 4 个错配的 2 个准种序列,而 ViSpA 则正确重建了最多有 2 个错配的 5 个准种序列,其中 2 个序列没有错配。ViSpA 的源代码可在 http://alla.cs.gsu.edu/~software/VISPA/vispa.html 获得。
ViSpA 能够从 454 焦磷酸测序读取中准确重建病毒准种谱。我们目前正在探索适用于细菌宏基因组样本和真核生物种群生态样本的高通量测序数据的分析扩展。