Chen Li, Xuan Jianhua, Riggins Rebecca B, Clarke Robert, Wang Yue
Department of Electrical and Computer Engineering, Virginia Polytechnic Institute and State University, Arlington, VA, USA.
BMC Syst Biol. 2011 Oct 12;5:161. doi: 10.1186/1752-0509-5-161.
One of the major goals in gene and protein expression profiling of cancer is to identify biomarkers and build classification models for prediction of disease prognosis or treatment response. Many traditional statistical methods, based on microarray gene expression data alone and individual genes' discriminatory power, often fail to identify biologically meaningful biomarkers thus resulting in poor prediction performance across data sets. Nonetheless, the variables in multivariable classifiers should synergistically interact to produce more effective classifiers than individual biomarkers.
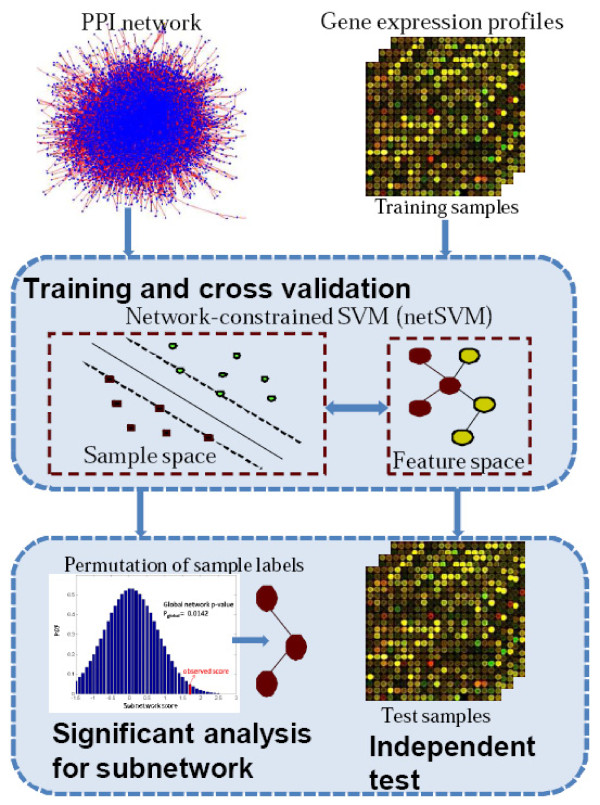
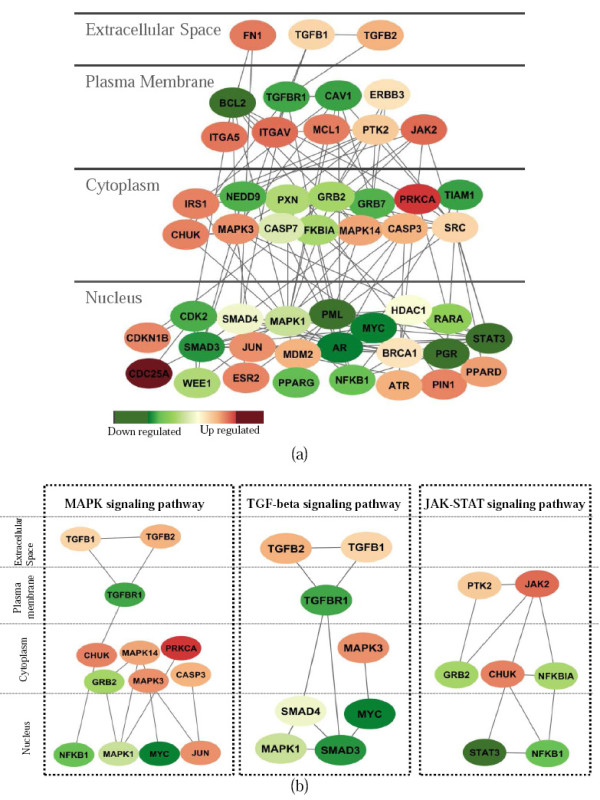
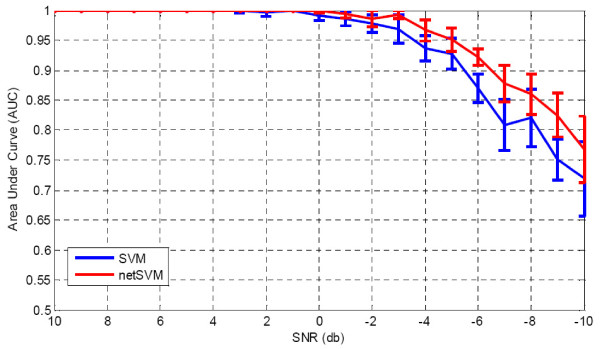
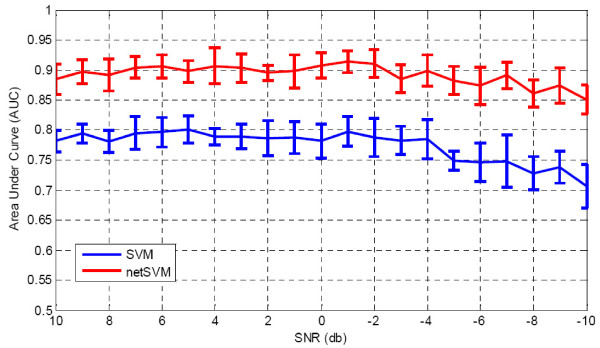
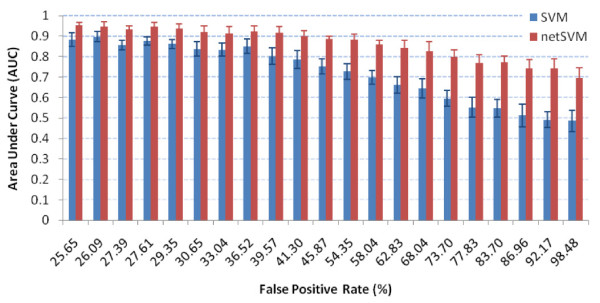
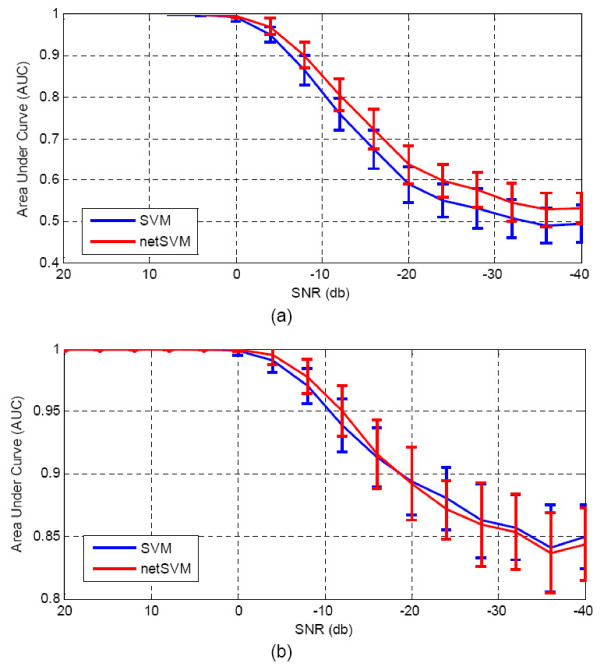
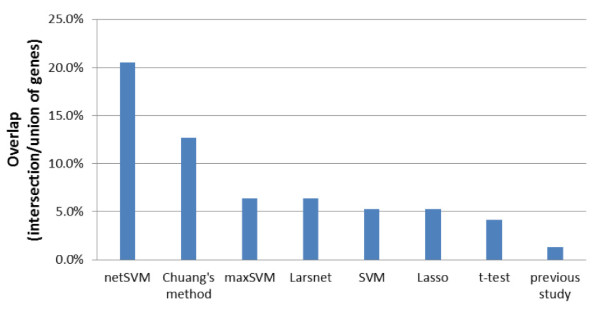
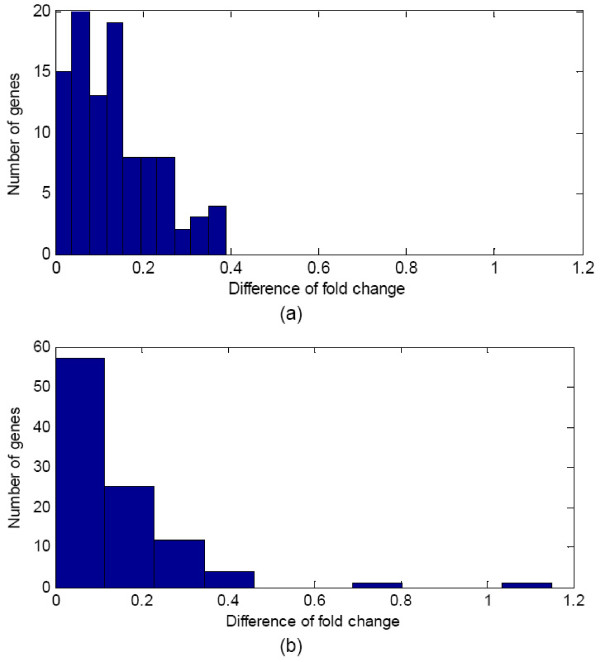
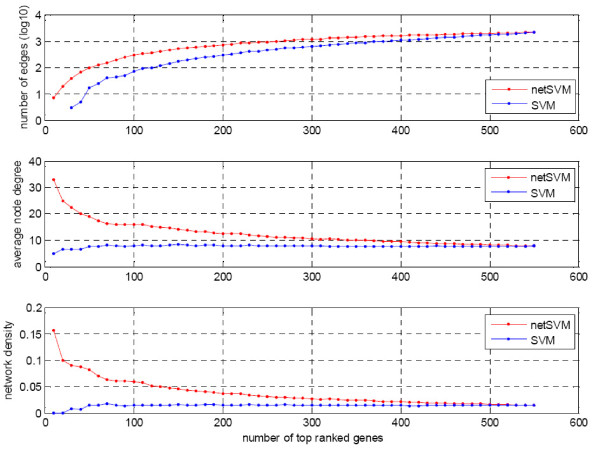
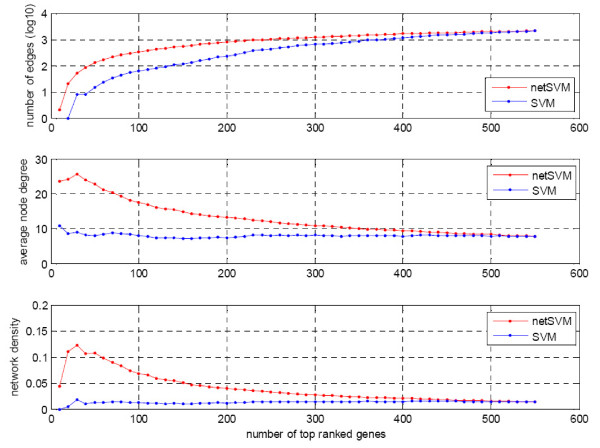
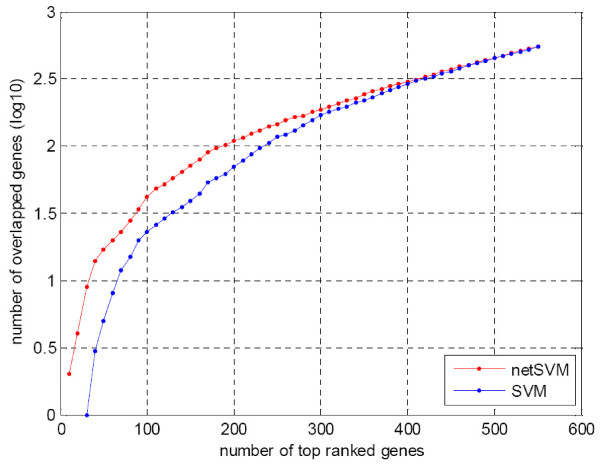
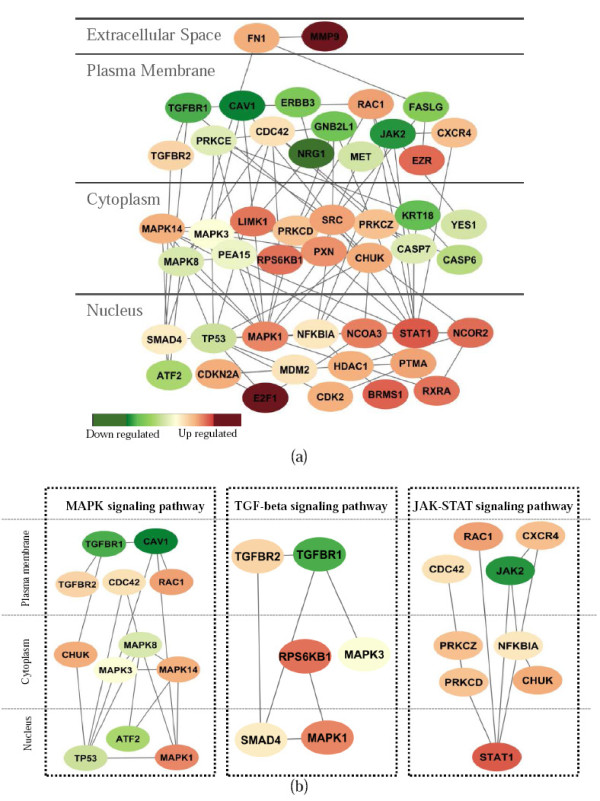
We developed an integrated approach, namely network-constrained support vector machine (netSVM), for cancer biomarker identification with an improved prediction performance. The netSVM approach is specifically designed for network biomarker identification by integrating gene expression data and protein-protein interaction data. We first evaluated the effectiveness of netSVM using simulation studies, demonstrating its improved performance over state-of-the-art network-based methods and gene-based methods for network biomarker identification. We then applied the netSVM approach to two breast cancer data sets to identify prognostic signatures for prediction of breast cancer metastasis. The experimental results show that: (1) network biomarkers identified by netSVM are highly enriched in biological pathways associated with cancer progression; (2) prediction performance is much improved when tested across different data sets. Specifically, many genes related to apoptosis, cell cycle, and cell proliferation, which are hallmark signatures of breast cancer metastasis, were identified by the netSVM approach. More importantly, several novel hub genes, biologically important with many interactions in PPI network but often showing little change in expression as compared with their downstream genes, were also identified as network biomarkers; the genes were enriched in signaling pathways such as TGF-beta signaling pathway, MAPK signaling pathway, and JAK-STAT signaling pathway. These signaling pathways may provide new insight to the underlying mechanism of breast cancer metastasis.
We have developed a network-based approach for cancer biomarker identification, netSVM, resulting in an improved prediction performance with network biomarkers. We have applied the netSVM approach to breast cancer gene expression data to predict metastasis in patients. Network biomarkers identified by netSVM reveal potential signaling pathways associated with breast cancer metastasis, and help improve the prediction performance across independent data sets.
癌症基因和蛋白质表达谱分析的主要目标之一是识别生物标志物并构建用于预测疾病预后或治疗反应的分类模型。许多传统统计方法仅基于微阵列基因表达数据和单个基因的判别能力,常常无法识别具有生物学意义的生物标志物,从而导致跨数据集的预测性能较差。尽管如此,多变量分类器中的变量应协同相互作用,以产生比单个生物标志物更有效的分类器。
我们开发了一种综合方法,即网络约束支持向量机(netSVM),用于癌症生物标志物识别,具有改进的预测性能。netSVM方法通过整合基因表达数据和蛋白质-蛋白质相互作用数据,专门设计用于网络生物标志物识别。我们首先使用模拟研究评估了netSVM的有效性,证明其在网络生物标志物识别方面比基于网络的现有方法和基于基因的方法具有更好的性能。然后,我们将netSVM方法应用于两个乳腺癌数据集,以识别用于预测乳腺癌转移的预后特征。实验结果表明:(1)netSVM识别的网络生物标志物在与癌症进展相关的生物途径中高度富集;(2)在不同数据集上进行测试时,预测性能有了很大提高。具体而言,netSVM方法识别出许多与细胞凋亡、细胞周期和细胞增殖相关的基因,这些是乳腺癌转移的标志性特征。更重要的是,几个新的枢纽基因也被识别为网络生物标志物,这些基因在蛋白质-蛋白质相互作用网络中有许多相互作用,在生物学上很重要,但与它们的下游基因相比,表达往往变化不大;这些基因在TGF-β信号通路、MAPK信号通路和JAK-STAT信号通路等信号通路中富集。这些信号通路可能为乳腺癌转移的潜在机制提供新的见解。
我们开发了一种基于网络的癌症生物标志物识别方法netSVM,通过网络生物标志物提高了预测性能。我们已将netSVM方法应用于乳腺癌基因表达数据,以预测患者的转移情况。netSVM识别的网络生物标志物揭示了与乳腺癌转移相关的潜在信号通路,并有助于提高跨独立数据集的预测性能。