Nutrition and Genomics Laboratory, Jean Mayer United States Department of Agriculture Human Nutrition Research Center on Aging at Tufts University, Boston, MA, USA.
BMC Genomics. 2011 Oct 13;12:504. doi: 10.1186/1471-2164-12-504.
Gene variants within regulatory regions are thought to be major contributors of the variation of complex traits/diseases. Genome wide association studies (GWAS), have identified scores of genetic variants that appear to contribute to human disease risk. However, most of these variants do not appear to be functional. Thus, the significance of the association may be brought up by still unknown mechanisms or by linkage disequilibrium (LD) with functional polymorphisms. In the present study, focused on functional variants related with the binding of microRNAs (miR), we utilized SNP data, including newly released 1000 Genomes Project data to perform a genome-wide scan of SNPs that abrogate or create miR recognition element (MRE) seed sites (MRESS).
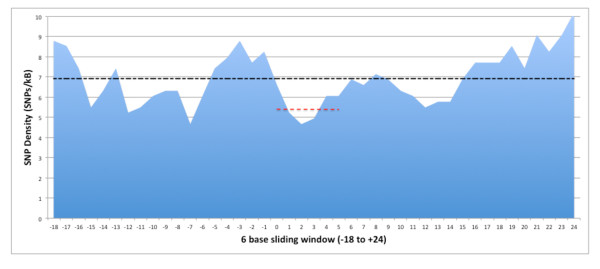
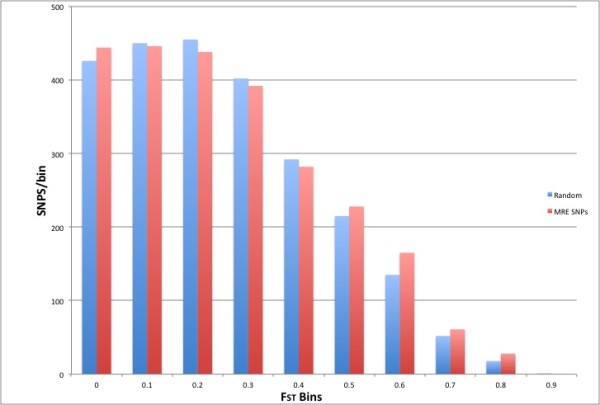
We identified 2723 SNPs disrupting, and 22295 SNPs creating MRESSs. We estimated the percent of SNPs falling within both validated (5%) and predicted conserved MRESSs (3%). We determined 87 of these MRESS SNPs were listed in GWAS association studies, or in strong LD with a GWAS SNP, and may represent the functional variants of identified GWAS SNPs. Furthermore, 39 of these have evidence of co-expression of target mRNA and the predicted miR. We also gathered previously published eQTL data supporting a functional role for four of these SNPs shown to associate with disease phenotypes. Comparison of FST statistics (a measure of population subdivision) for predicted MRESS SNPs against non MRESS SNPs revealed a significantly higher (P = 0.0004) degree of subdivision among MRESS SNPs, suggesting a role for these SNPs in environmentally driven selection.
We have demonstrated the potential of publicly available resources to identify high priority candidate SNPs for functional studies and for disease risk prediction.
调控区域内的基因变异被认为是复杂性状/疾病变异的主要因素。全基因组关联研究(GWAS)已经确定了许多遗传变异,这些变异似乎与人类疾病风险有关。然而,这些变异中的大多数似乎并不具有功能。因此,关联的意义可能是由未知的机制或与功能性多态性的连锁不平衡(LD)引起的。在本研究中,我们专注于与 microRNAs(miR)结合相关的功能性变异,利用包括新发布的 1000 基因组计划数据在内的 SNP 数据,进行了全基因组范围内的 SNP 扫描,这些 SNP 会破坏或创建 miR 识别元件(MRE)种子位点(MRESS)。
我们鉴定了 2723 个破坏 MRESS 的 SNP 和 22295 个创建 MRESS 的 SNP。我们估计了落在验证(5%)和预测保守 MRESS(3%)内的 SNP 的比例。我们确定了这些 MRESS SNP 中有 87 个在 GWAS 关联研究中列出,或者与 GWAS SNP 强连锁,可能代表已识别的 GWAS SNP 的功能性变异。此外,其中 39 个具有目标 mRNA 和预测 miR 共表达的证据。我们还收集了先前发表的 eQTL 数据,支持其中四个与疾病表型相关的 SNP 具有功能性作用。与非 MRESS SNP 相比,预测 MRESS SNP 的 FST 统计量(衡量种群划分的指标)的比较显示,MRESS SNP 之间的划分程度显著更高(P=0.0004),这表明这些 SNP 在环境驱动的选择中发挥了作用。
我们已经证明了利用公共资源识别高优先级候选 SNP 进行功能研究和疾病风险预测的潜力。