Centre for Molecular Medicine and Therapeutics, Child and Family Research Institute, University of British Columbia, Vancouver, British Columbia, Canada.
BMC Med Genomics. 2014 Jun 11;7:34. doi: 10.1186/1755-8794-7-34.
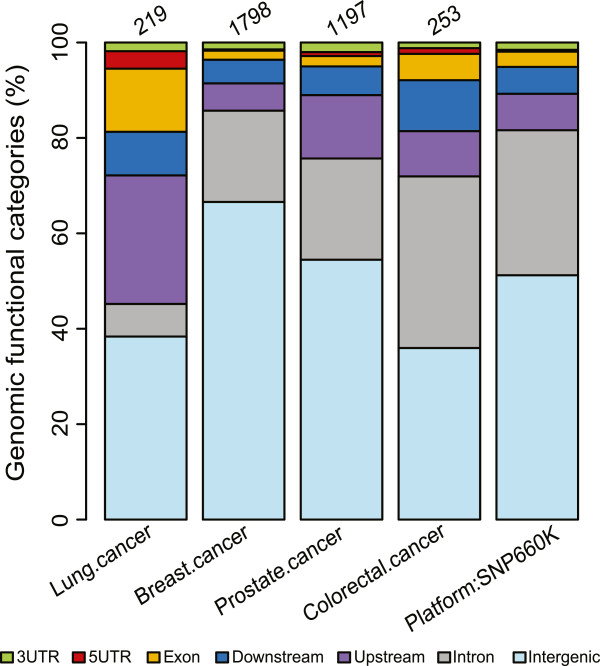
Genome wide association studies (GWAS) are a population-scale approach to the identification of segments of the genome in which genetic variations may contribute to disease risk. Current methods focus on the discovery of single nucleotide polymorphisms (SNPs) associated with disease traits. As there are many SNPs within identified risk loci, and the majority of these are situated within non-coding regions, a key challenge is to identify and prioritize variants affecting regulatory sequences that are likely to contribute to the phenotype assessed.
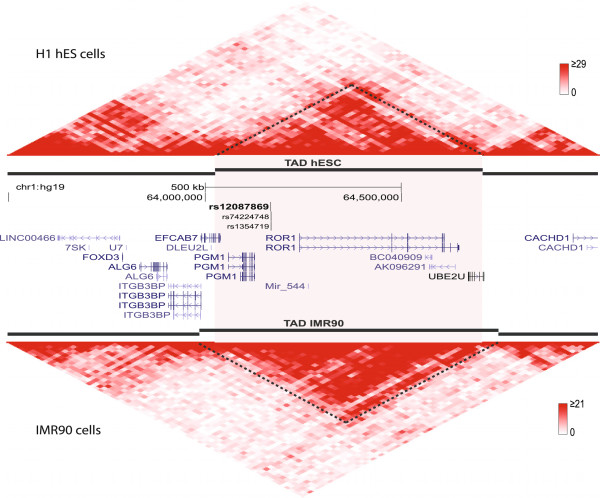
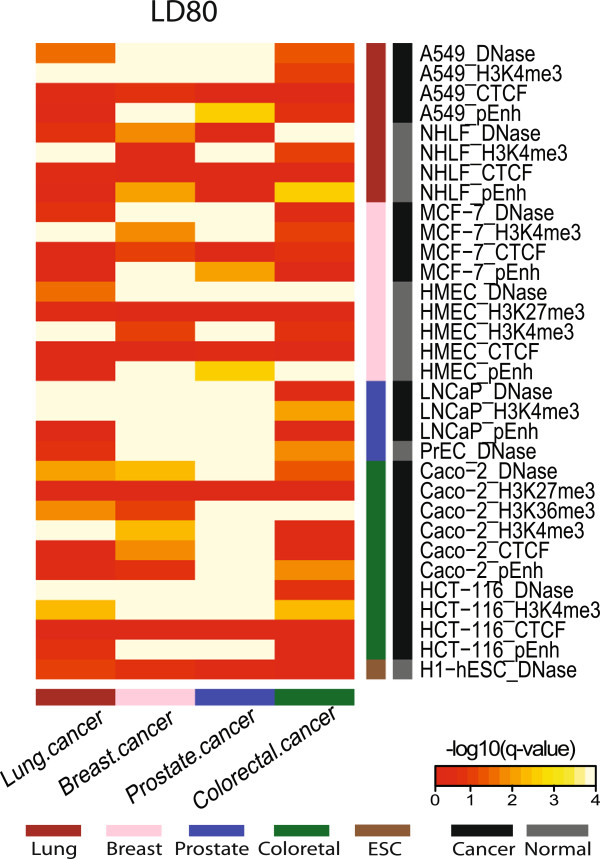
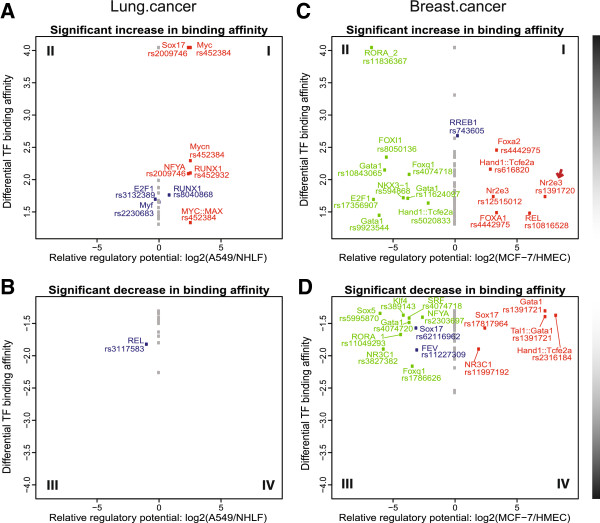
We focused investigation on SNPs within lung and breast cancer GWAS loci that reached genome-wide significance for potential roles in gene regulation with a specific focus on SNPs likely to disrupt transcription factor binding sites. Within risk loci, the regulatory potential of sub-regions was classified using relevant open chromatin and epigenetic high throughput sequencing data sets from the ENCODE project in available cancer and normal cell lines. Furthermore, transcription factor affinity altering variants were predicted by comparison of position weight matrix scores between disease and reference alleles. Lastly, ChIP-seq data of transcription associated factors and topological domains were included as binding evidence and potential gene target inference.
The sets of SNPs, including both the disease-associated markers and those in high linkage disequilibrium with them, were significantly over-represented in regulatory sequences of cancer and/or normal cells; however, over-representation was generally not restricted to disease-relevant tissue specific regions. The calculated regulatory potential, allelic binding affinity scores and ChIP-seq binding evidence were the three criteria used to prioritize candidates. Fitting all three criteria, we highlighted breast cancer susceptibility SNPs and a borderline lung cancer relevant SNP located in cancer-specific enhancers overlapping multiple distinct transcription associated factor ChIP-seq binding sites.
Incorporating high throughput sequencing epigenetic and transcription factor data sets from both cancer and normal cells into cancer genetic studies reveals potential functional SNPs and informs subsequent characterization efforts.
全基因组关联研究(GWAS)是一种针对基因组中可能导致疾病风险的遗传变异片段进行鉴定的群体研究方法。目前的方法主要侧重于发现与疾病特征相关的单核苷酸多态性(SNP)。由于在已确定的风险基因座内存在许多 SNP,而且大多数 SNP 位于非编码区域内,因此一个关键的挑战是识别和优先考虑可能影响受评估表型的调节序列的变体。
我们集中研究了肺癌和乳腺癌 GWAS 基因座内的 SNP,这些 SNP 在基因调控方面具有全基因组意义,特别是那些可能破坏转录因子结合位点的 SNP。在风险基因座内,使用 ENCODE 项目中来自可用癌症和正常细胞系的相关开放染色质和表观遗传高通量测序数据集,对亚区域的调节潜力进行分类。此外,通过比较疾病和参考等位基因之间的位置权重矩阵得分,预测转录因子亲和力改变的变体。最后,将与转录相关的因子的 ChIP-seq 数据和拓扑结构域纳入作为结合证据和潜在的基因靶标推断。
包括疾病相关标记物和与它们高度连锁不平衡的 SNP 在内的 SNP 集在癌症和/或正常细胞的调节序列中显著过表达;然而,过表达通常不限于与疾病相关的组织特异性区域。计算的调节潜力、等位结合亲和力得分和 ChIP-seq 结合证据是用于优先考虑候选物的三个标准。符合所有三个标准,我们强调了乳腺癌易感性 SNP 和一个位于重叠多个不同转录相关因子 ChIP-seq 结合位点的癌症特异性增强子中的肺癌相关 SNP。
将来自癌症和正常细胞的高通量测序表观遗传和转录因子数据集纳入癌症遗传研究中,可以揭示潜在的功能 SNP,并为随后的特征描述工作提供信息。