Bayer AG, Bayer Technology Services, 51368 Leverkusen, Germany.
BMC Med Genomics. 2011 Oct 14;4:73. doi: 10.1186/1755-8794-4-73.
Identifying stable gene lists for diagnosis, prognosis prediction, and treatment guidance of tumors remains a major challenge in cancer research. Microarrays measuring differential gene expression are widely used and should be versatile predictors of disease and other phenotypic data. However, gene expression profile studies and predictive biomarkers are often of low power, requiring numerous samples for a sound statistic, or vary between studies. Given the inconsistency of results across similar studies, methods that identify robust biomarkers from microarray data are needed to relay true biological information. Here we present a method to demonstrate that gene list stability and predictive power depends not only on the size of studies, but also on the clinical phenotype.
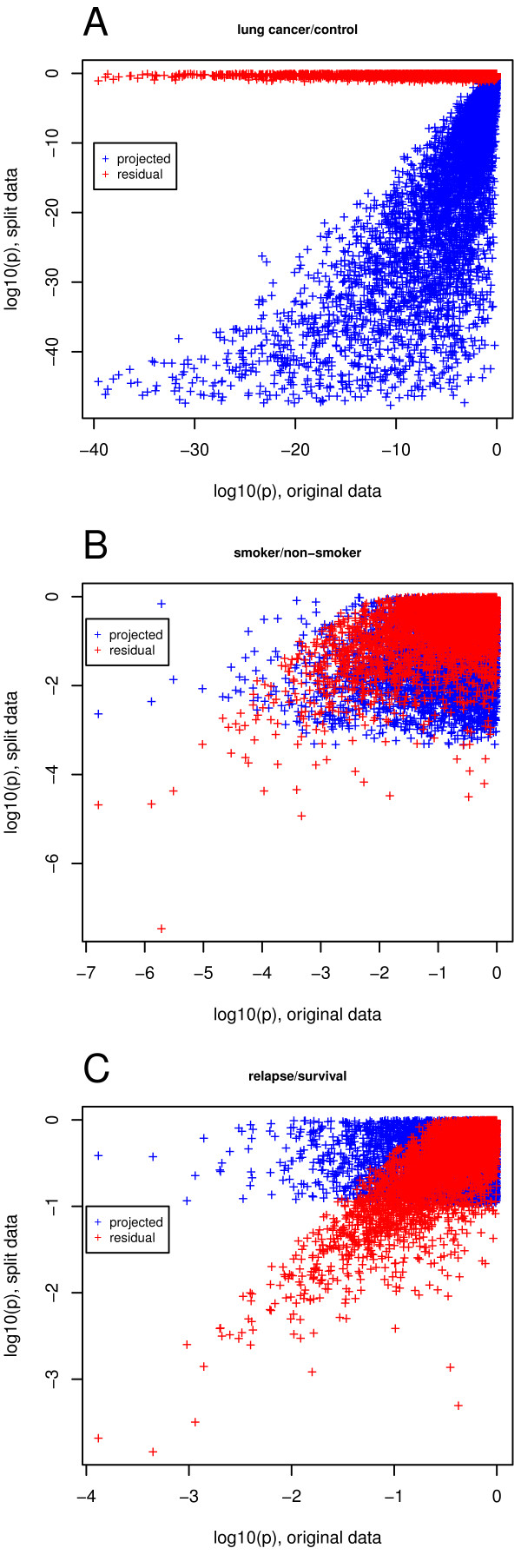
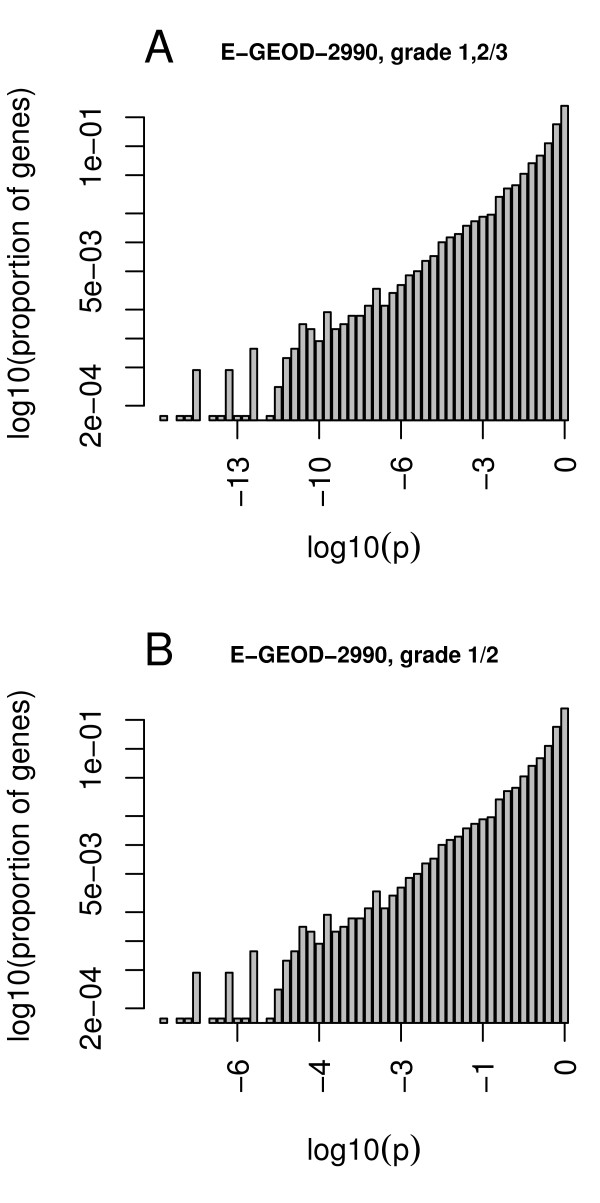
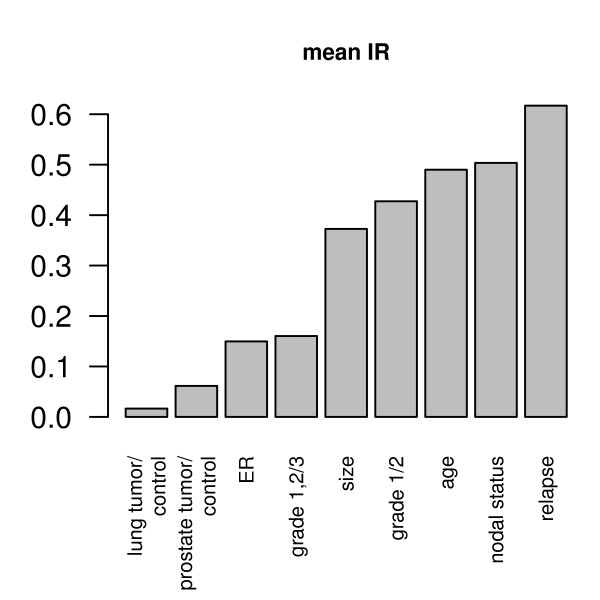
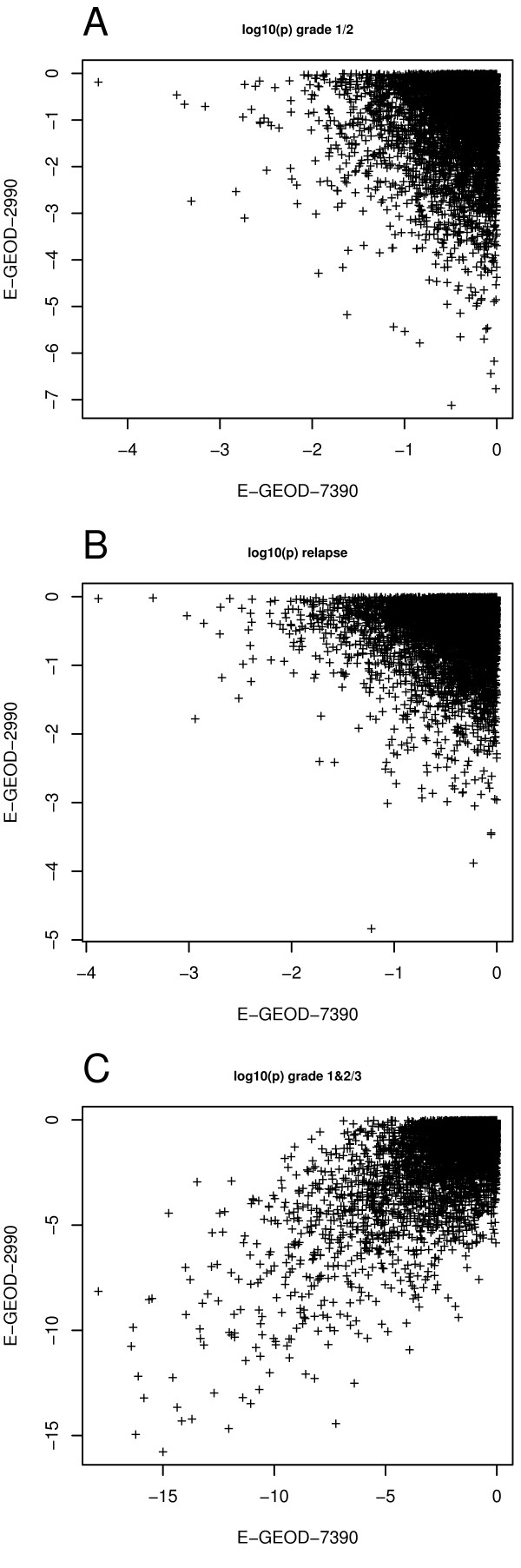
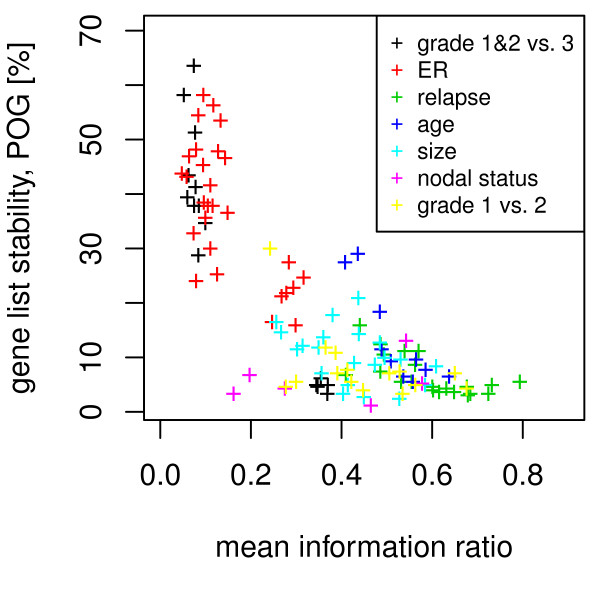
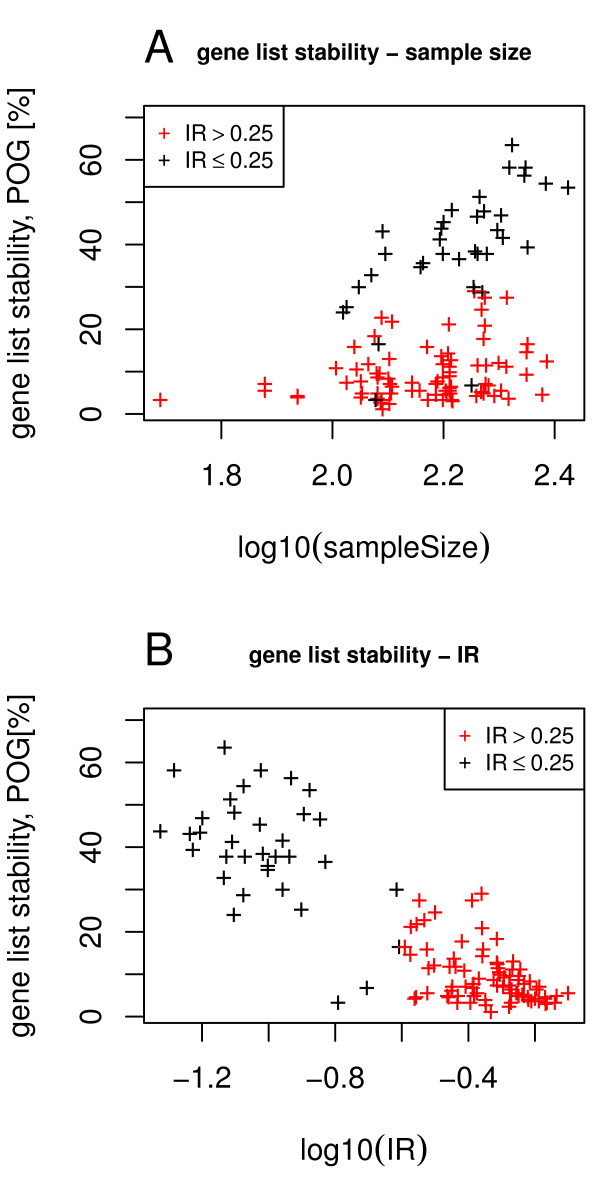
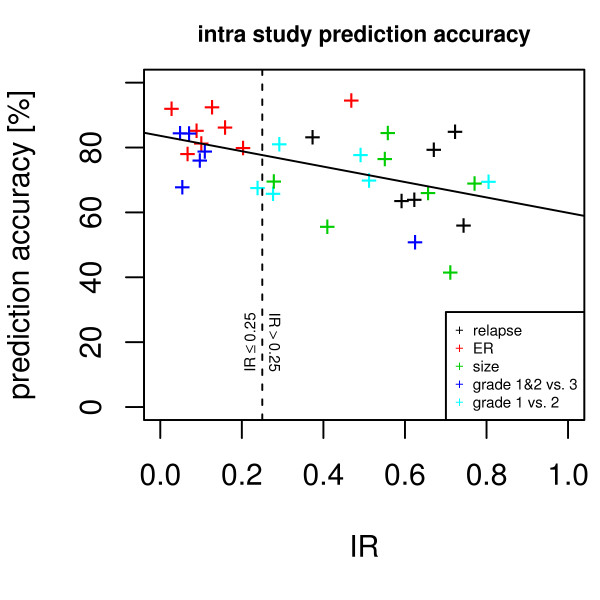
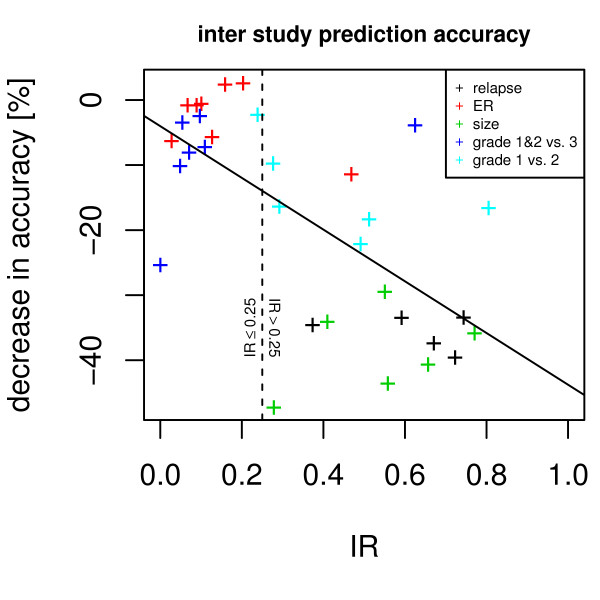
Our method projects genomic tumor expression data to a lower dimensional space representing the main variation in the data. Some information regarding the phenotype resides in this low dimensional space, while some information resides in the residuum. We then introduce an information ratio (IR) as a metric defined by the partition between projected and residual space. Upon grouping phenotypes such as tumor tissue, histological grades, relapse, or aging, we show that higher IR values correlated with phenotypes that yield less robust biomarkers whereas lower IR values showed higher transferability across studies. Our results indicate that the IR is correlated with predictive accuracy. When tested across different published datasets, the IR can identify information-rich data characterizing clinical phenotypes and stable biomarkers.
The IR presents a quantitative metric to estimate the information content of gene expression data with respect to particular phenotypes.
鉴定用于肿瘤诊断、预后预测和治疗指导的稳定基因列表仍然是癌症研究中的一个主要挑战。测量差异基因表达的微阵列被广泛应用,并且应该是疾病和其他表型数据的多功能预测因子。然而,基因表达谱研究和预测生物标志物的功效往往较低,需要大量样本进行稳健的统计分析,或者在研究之间存在差异。鉴于类似研究的结果不一致,需要有从微阵列数据中识别稳健生物标志物的方法来传递真实的生物学信息。在这里,我们提出了一种方法来证明基因列表的稳定性和预测能力不仅取决于研究的规模,还取决于临床表型。
我们的方法将基因组肿瘤表达数据投影到一个较低维度的空间中,代表数据的主要变化。一些关于表型的信息驻留在这个低维空间中,而一些信息驻留在残差中。然后,我们引入了一个信息比(IR)作为一个度量标准,由投影空间和残差空间之间的划分定义。在对肿瘤组织、组织学分级、复发或老化等表型进行分组后,我们表明,较高的 IR 值与产生不稳健生物标志物的表型相关,而较低的 IR 值则表现出较高的跨研究可转移性。我们的结果表明,IR 与预测准确性相关。当在不同的已发表数据集上进行测试时,IR 可以识别出以临床表型和稳定生物标志物为特征的富含信息的数据。
IR 提供了一种定量度量标准,可以估计特定表型的基因表达数据的信息量。