Max Delbrück Center for Molecular Medicine, Berlin, Germany.
PLoS One. 2011;6(10):e26069. doi: 10.1371/journal.pone.0026069. Epub 2011 Oct 10.
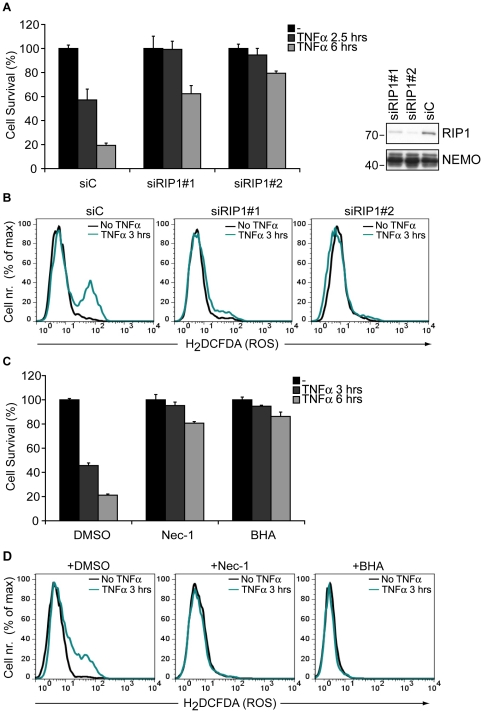
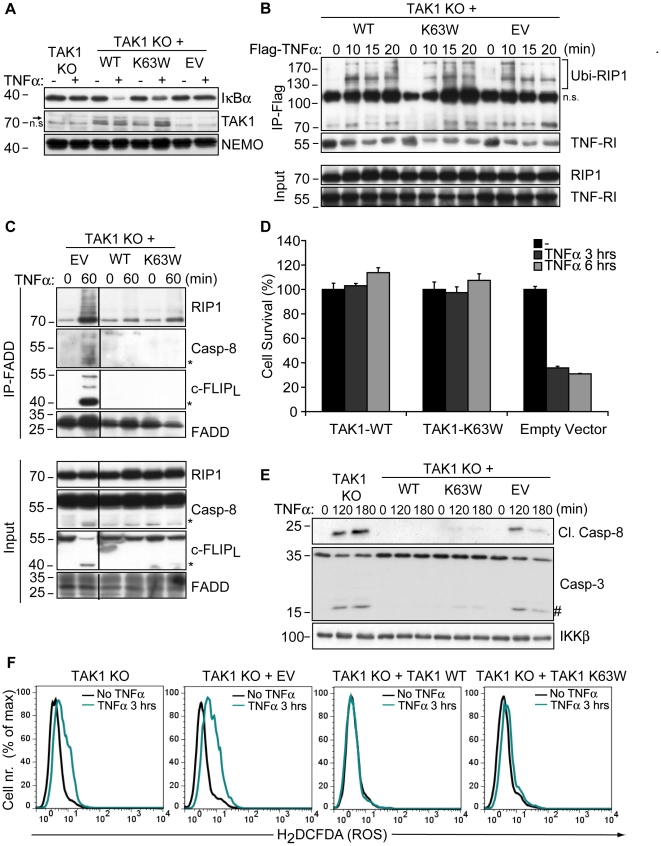
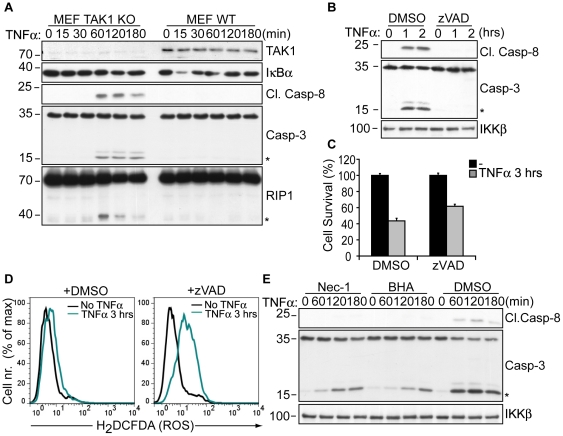
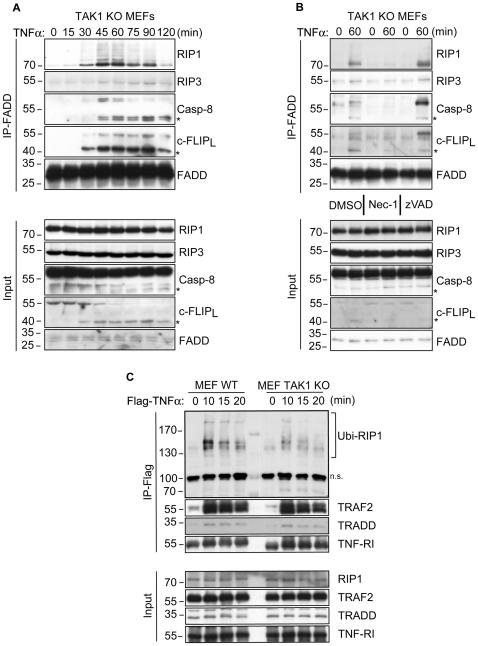
Death receptor-induced programmed necrosis is regarded as a secondary death mechanism dominating only in cells that cannot properly induce caspase-dependent apoptosis. Here, we show that in cells lacking TGFβ-activated Kinase-1 (TAK1) expression, catalytically active Receptor Interacting Protein 1 (RIP1)-dependent programmed necrosis overrides apoptotic processes following Tumor Necrosis Factor-α (TNFα) stimulation and results in rapid cell death. Importantly, the activation of the caspase cascade and caspase-8-mediated RIP1 cleavage in TNFα-stimulated TAK1 deficient cells is not sufficient to prevent RIP1-dependent necrosome formation and subsequent programmed necrosis. Our results demonstrate that TAK1 acts independently of its kinase activity to prevent the premature dissociation of ubiquitinated-RIP1 from TNFα-stimulated TNF-receptor I and also to inhibit the formation of TNFα-induced necrosome complex consisting of RIP1, RIP3, FADD, caspase-8 and cFLIP(L). The surprising prevalence of catalytically active RIP1-dependent programmed necrosis over apoptosis despite ongoing caspase activity implicates a complex regulatory mechanism governing the decision between both cell death pathways following death receptor stimulation.
死亡受体诱导的程序性细胞坏死被认为是一种次要的死亡机制,仅在不能正确诱导半胱天冬酶依赖性细胞凋亡的细胞中起作用。在这里,我们表明在缺乏 TGFβ 激活激酶 1(TAK1)表达的细胞中,催化活性的受体相互作用蛋白 1(RIP1)依赖性程序性细胞坏死会在肿瘤坏死因子-α(TNFα)刺激后取代凋亡过程,并导致快速细胞死亡。重要的是,在 TNFα 刺激的 TAK1 缺陷细胞中,胱天蛋白酶级联的激活和胱天蛋白酶-8 介导的 RIP1 裂解不足以防止 RIP1 依赖性坏死体形成和随后的程序性细胞坏死。我们的研究结果表明,TAK1 独立于其激酶活性发挥作用,以防止泛素化的 RIP1 从 TNFα 刺激的 TNF 受体 I 过早解离,并抑制由 RIP1、RIP3、FADD、胱天蛋白酶-8 和 cFLIP(L)组成的 TNFα 诱导的坏死体复合物的形成。尽管存在持续的胱天蛋白酶活性,但催化活性的 RIP1 依赖性程序性细胞坏死对细胞凋亡的普遍性,暗示了在死亡受体刺激后,两种细胞死亡途径之间的决策存在复杂的调控机制。