Department of Environmental Science, Policy, and Management, University of California, Berkeley, California, United States of America.
PLoS Comput Biol. 2011 Oct;7(10):e1002230. doi: 10.1371/journal.pcbi.1002230. Epub 2011 Oct 20.
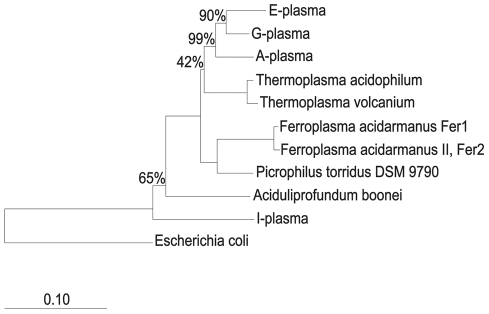
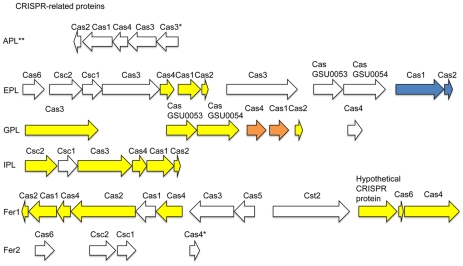
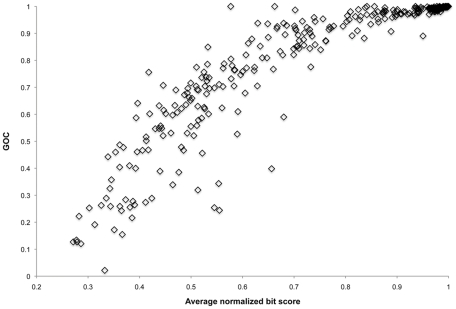
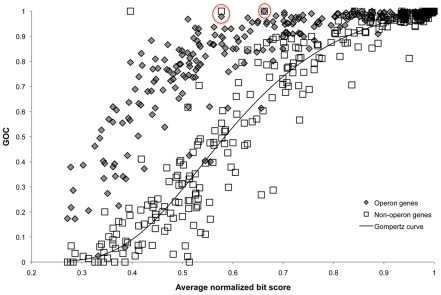
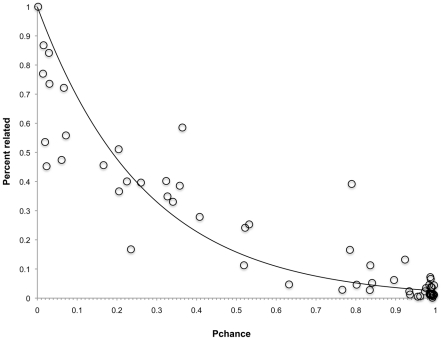
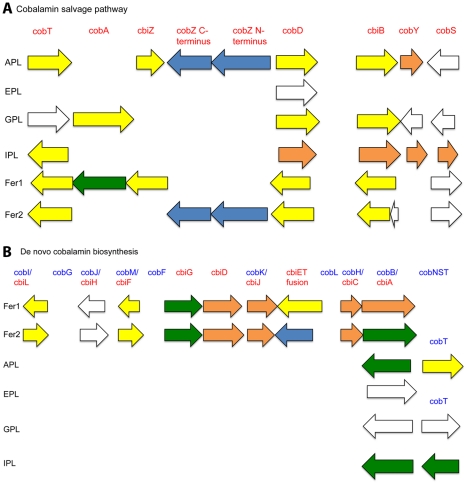
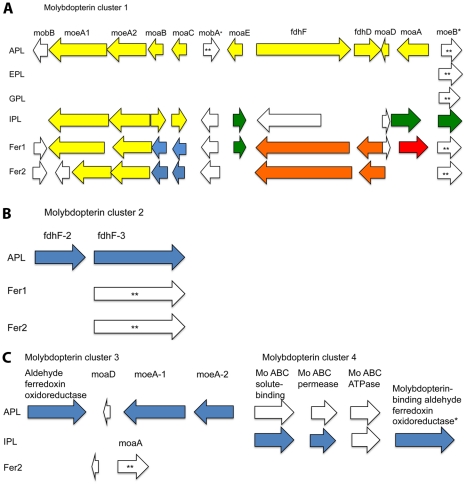
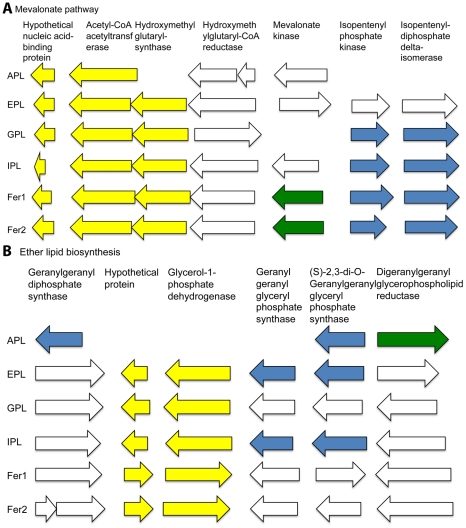
During microbial evolution, genome rearrangement increases with increasing sequence divergence. If the relationship between synteny and sequence divergence can be modeled, gene clusters in genomes of distantly related organisms exhibiting anomalous synteny can be identified and used to infer functional conservation. We applied the phylogenetic pairwise comparison method to establish and model a strong correlation between synteny and sequence divergence in all 634 available Archaeal and Bacterial genomes from the NCBI database and four newly assembled genomes of uncultivated Archaea from an acid mine drainage (AMD) community. In parallel, we established and modeled the trend between synteny and functional relatedness in the 118 genomes available in the STRING database. By combining these models, we developed a gene functional annotation method that weights evolutionary distance to estimate the probability of functional associations of syntenous proteins between genome pairs. The method was applied to the hypothetical proteins and poorly annotated genes in newly assembled acid mine drainage Archaeal genomes to add or improve gene annotations. This is the first method to assign possible functions to poorly annotated genes through quantification of the probability of gene functional relationships based on synteny at a significant evolutionary distance, and has the potential for broad application.
在微生物进化过程中,基因组重排随着序列分歧的增加而增加。如果可以对同线性与序列分歧之间的关系进行建模,那么就可以识别出在序列分歧较大的生物体基因组中表现出异常同线性的基因簇,并用于推断功能保守性。我们应用系统发育成对比较的方法,在来自 NCBI 数据库的 634 个可获得的古细菌和细菌基因组以及来自酸性矿山排水(AMD)群落的四个新组装的未培养古细菌基因组中建立并对同线性与序列分歧之间的强相关性进行建模。同时,我们在 STRING 数据库中 118 个可获得的基因组中建立并对同线性与功能相关性之间的趋势进行建模。通过组合这些模型,我们开发了一种基因功能注释方法,该方法根据进化距离为基因组对之间同线性蛋白的功能关联概率加权。该方法应用于新组装的酸性矿山排水古细菌基因组中的假设蛋白和注释较差的基因,以添加或改进基因注释。这是第一个通过基于同线性在显著进化距离上量化基因功能关系的概率来为注释较差的基因分配可能功能的方法,具有广泛的应用潜力。