Raes Jeroen, Korbel Jan O, Lercher Martin J, von Mering Christian, Bork Peer
European Molecular Biology Laboratory, Meyerhofstrasse 1, D-69117 Heidelberg, Germany.
Genome Biol. 2007;8(1):R10. doi: 10.1186/gb-2007-8-1-r10.
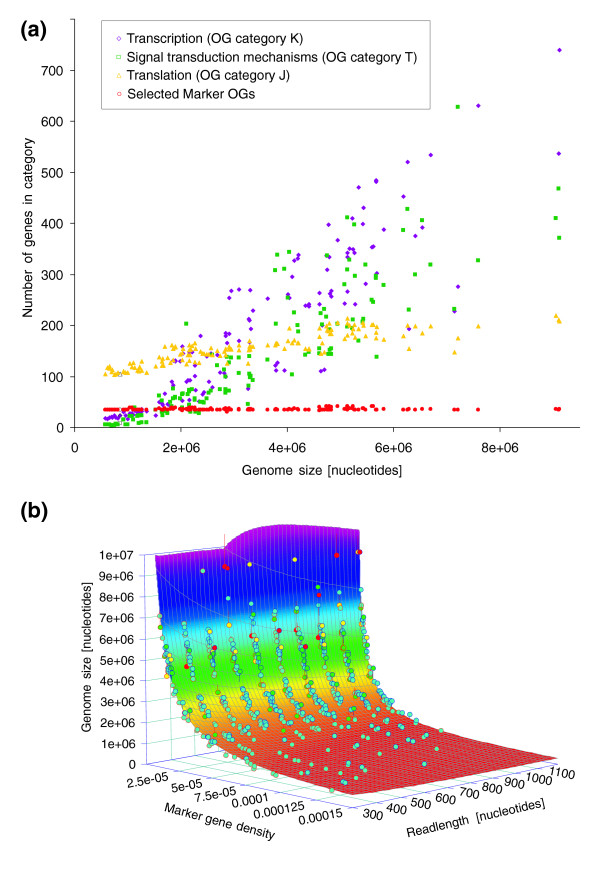
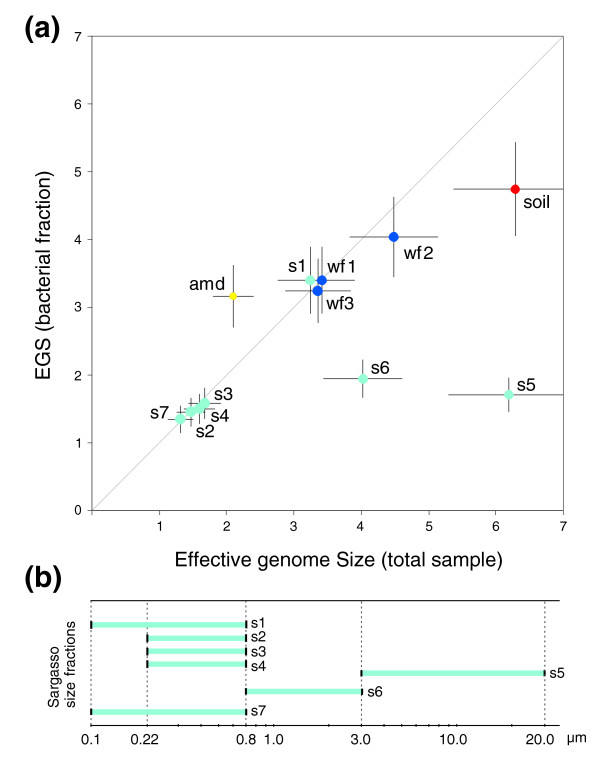
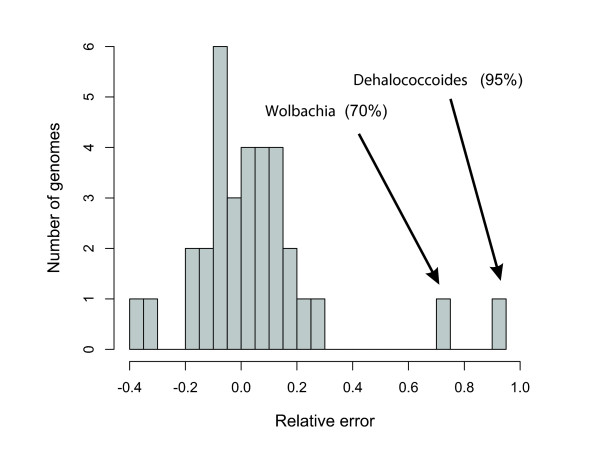
We introduce a novel computational approach to predict effective genome size (EGS; a measure that includes multiple plasmid copies, inserted sequences, and associated phages and viruses) from short sequencing reads of environmental genomics (or metagenomics) projects. We observe considerable EGS differences between environments and link this with ecologic complexity as well as species composition (for instance, the presence of eukaryotes). For example, we estimate EGS in a complex, organism-dense farm soil sample at about 6.3 megabases (Mb) whereas that of the bacteria therein is only 4.7 Mb; for bacteria in a nutrient-poor, organism-sparse ocean surface water sample, EGS is as low as 1.6 Mb. The method also permits evaluation of completion status and assembly bias in single-genome sequencing projects.
我们引入了一种新颖的计算方法,可根据环境基因组学(或宏基因组学)项目的短测序读数预测有效基因组大小(EGS,一种包含多个质粒拷贝、插入序列以及相关噬菌体和病毒的度量)。我们观察到不同环境之间的EGS存在显著差异,并将其与生态复杂性以及物种组成(例如真核生物的存在)联系起来。例如,我们估计在一个复杂、生物密集的农田土壤样本中,EGS约为630万个碱基对(Mb),而其中细菌的EGS仅为470万个碱基对;对于贫营养、生物稀疏的海洋表层水样本中的细菌,EGS低至160万个碱基对。该方法还允许评估单基因组测序项目的完成状态和组装偏差。