Department of Computer Science, Rutgers University, 110 Frelinghuysen Road, Piscataway, NJ 08854, USA.
BMC Bioinformatics. 2011 Nov 2;12:428. doi: 10.1186/1471-2105-12-428.
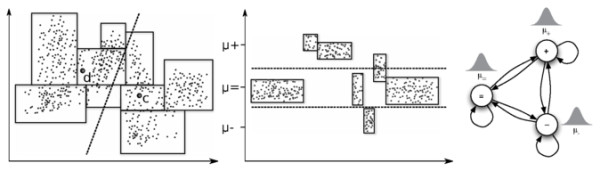
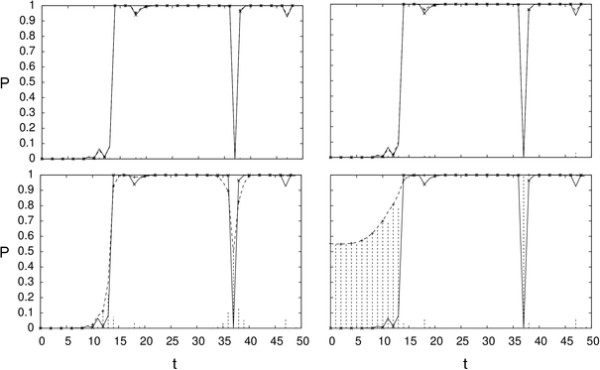
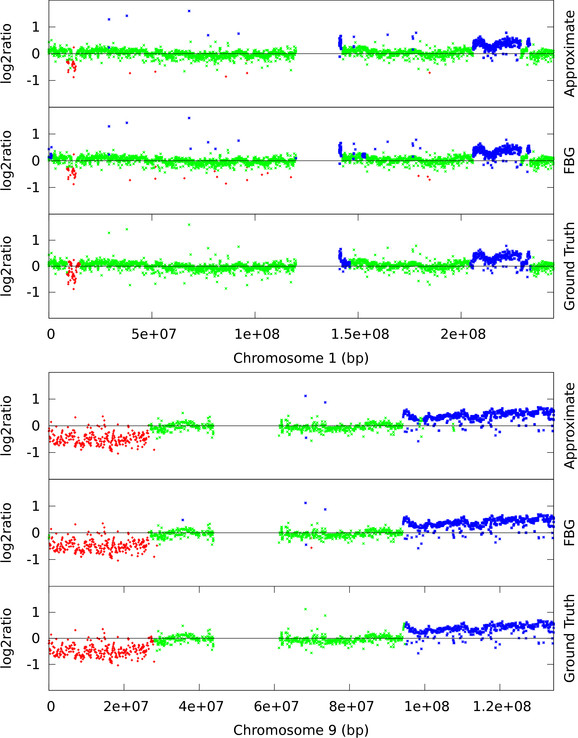
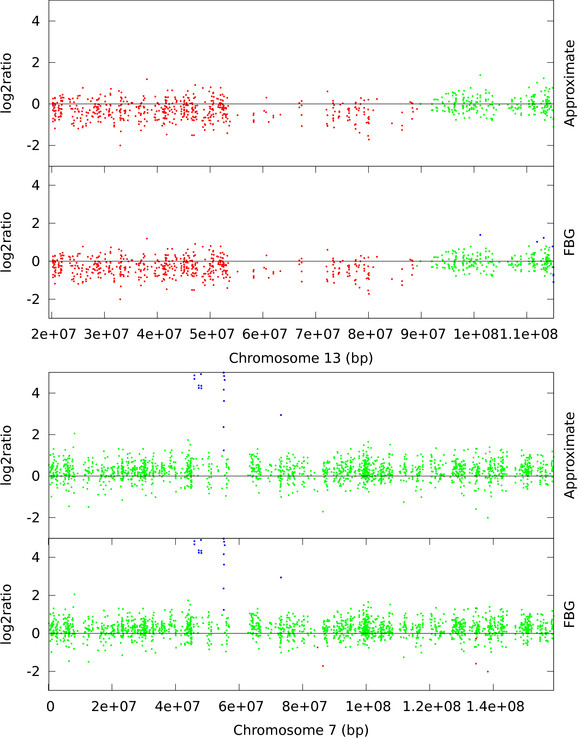
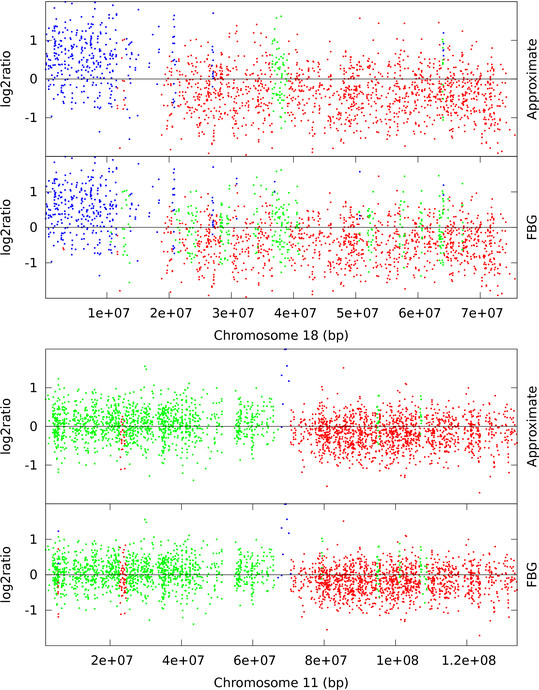
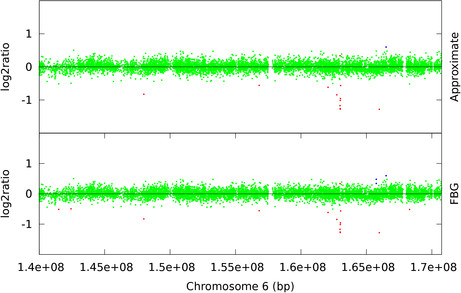
Hidden Markov Models (HMM) are often used for analyzing Comparative Genomic Hybridization (CGH) data to identify chromosomal aberrations or copy number variations by segmenting observation sequences. For efficiency reasons the parameters of a HMM are often estimated with maximum likelihood and a segmentation is obtained with the Viterbi algorithm. This introduces considerable uncertainty in the segmentation, which can be avoided with Bayesian approaches integrating out parameters using Markov Chain Monte Carlo (MCMC) sampling. While the advantages of Bayesian approaches have been clearly demonstrated, the likelihood based approaches are still preferred in practice for their lower running times; datasets coming from high-density arrays and next generation sequencing amplify these problems.
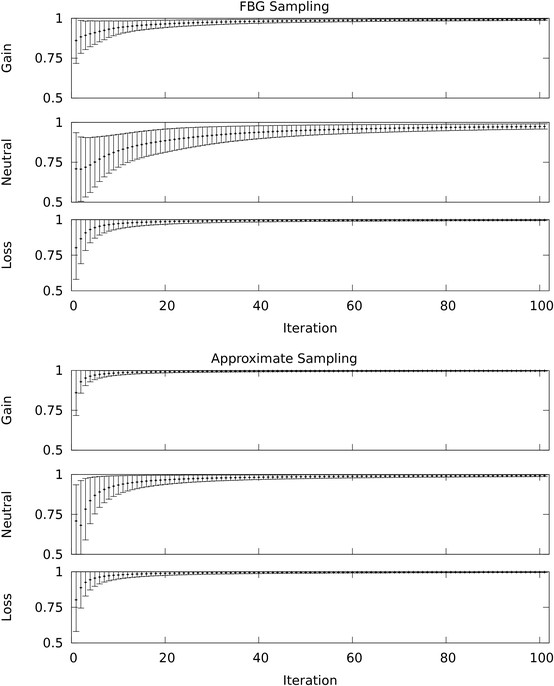
We propose an approximate sampling technique, inspired by compression of discrete sequences in HMM computations and by kd-trees to leverage spatial relations between data points in typical data sets, to speed up the MCMC sampling.
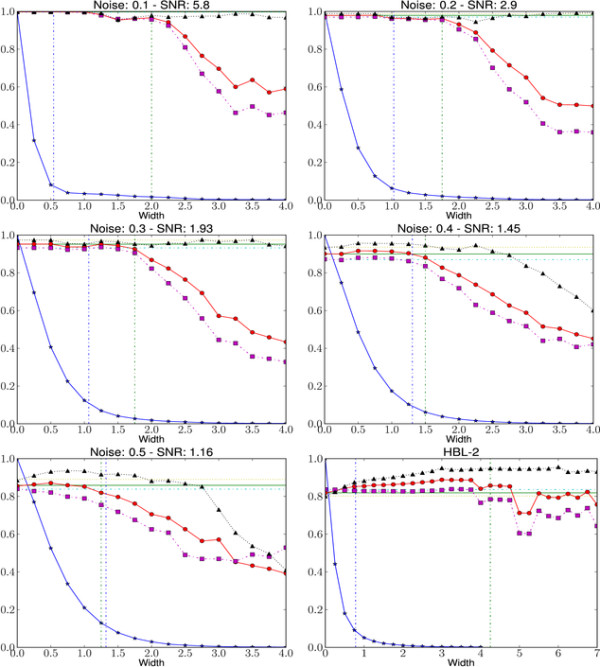
We test our approximate sampling method on simulated and biological ArrayCGH datasets and high-density SNP arrays, and demonstrate a speed-up of 10 to 60 respectively 90 while achieving competitive results with the state-of-the art Bayesian approaches.
An implementation of our method will be made available as part of the open source GHMM library from http://ghmm.org.
隐马尔可夫模型(HMM)常用于分析比较基因组杂交(CGH)数据,通过对观测序列进行分割来识别染色体异常或拷贝数变异。出于效率原因,HMM 的参数通常通过最大似然法进行估计,并通过维特比算法获得分割。这会在分割中引入相当大的不确定性,可以通过使用马尔可夫链蒙特卡罗(MCMC)采样来对参数进行贝叶斯推断的方法来避免。虽然贝叶斯方法的优势已得到明确证明,但由于其运行时间较短,基于似然的方法在实践中仍然更受欢迎;来自高密度阵列和下一代测序的数据集放大了这些问题。
我们提出了一种近似采样技术,该技术的灵感来自于 HMM 计算中离散序列的压缩和 kd-树,以利用典型数据集中数据点之间的空间关系,从而加快 MCMC 采样速度。
我们在模拟和生物 ArrayCGH 数据集以及高密度 SNP 阵列上测试了我们的近似采样方法,并分别实现了 10 到 60 倍和 90 倍的加速,同时与最先进的贝叶斯方法相比取得了有竞争力的结果。
我们的方法的实现将作为来自 http://ghmm.org 的开源 GHMM 库的一部分提供。