Department of Genetics, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
BMC Bioinformatics. 2010 Oct 31;11:539. doi: 10.1186/1471-2105-11-539.
Copy number variants (CNVs) have been demonstrated to occur at a high frequency and are now widely believed to make a significant contribution to the phenotypic variation in human populations. Array-based comparative genomic hybridization (array-CGH) and newly developed read-depth approach through ultrahigh throughput genomic sequencing both provide rapid, robust, and comprehensive methods to identify CNVs on a whole-genome scale.
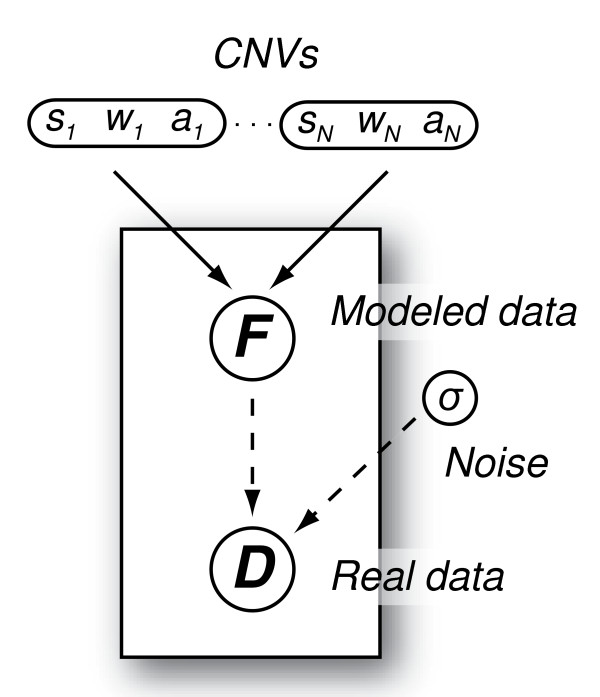
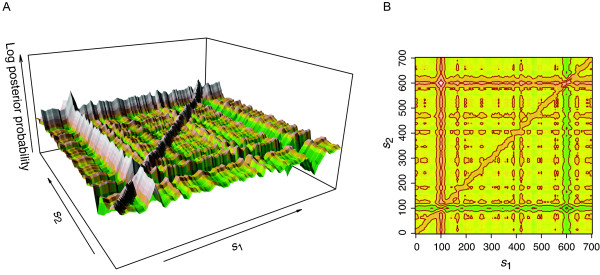
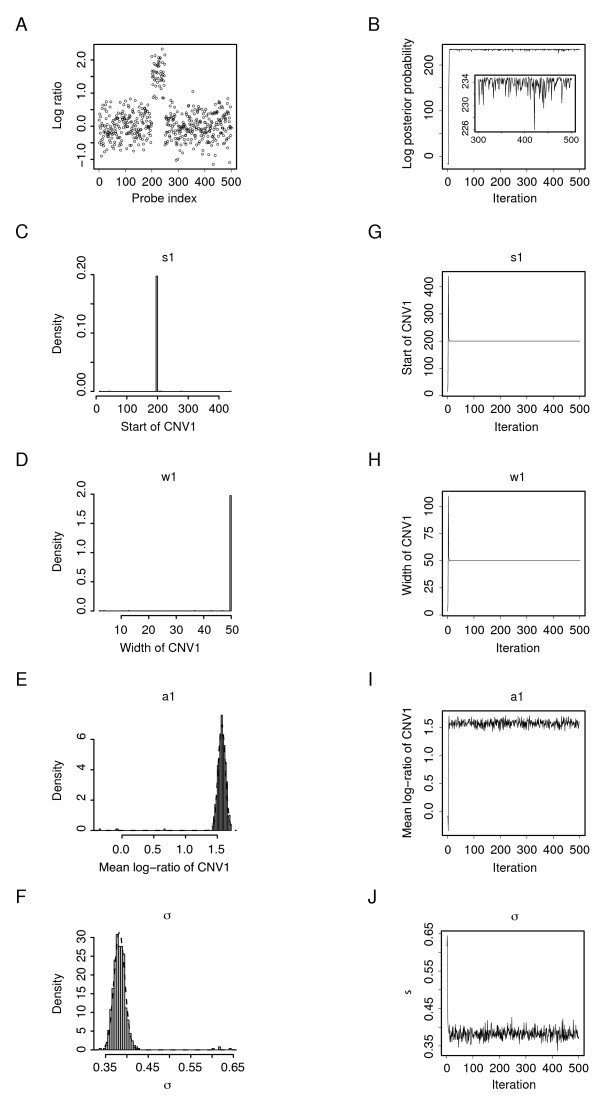
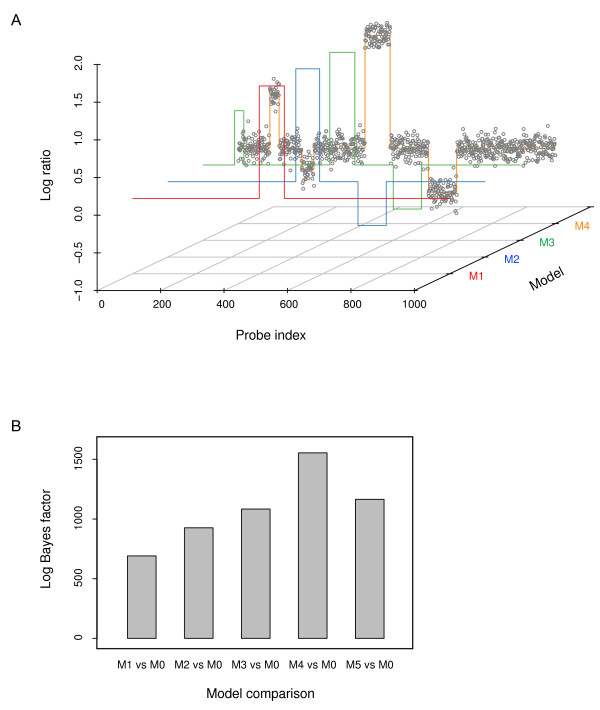
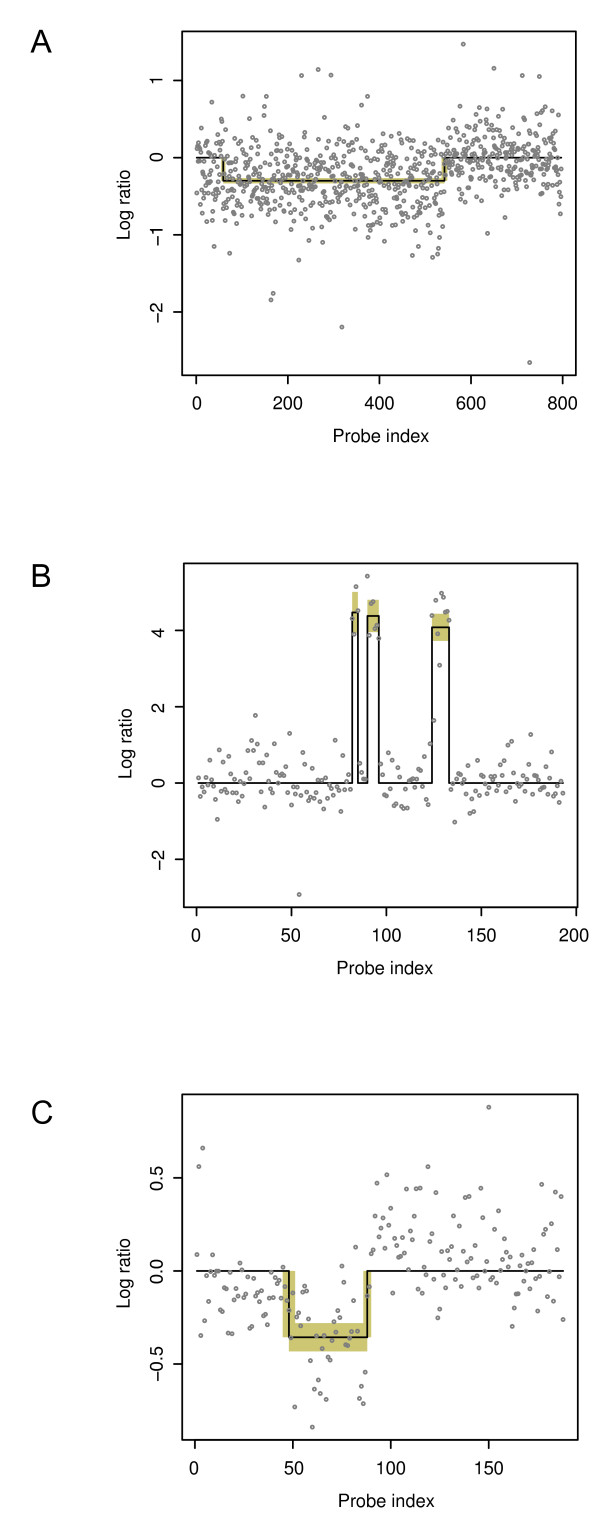
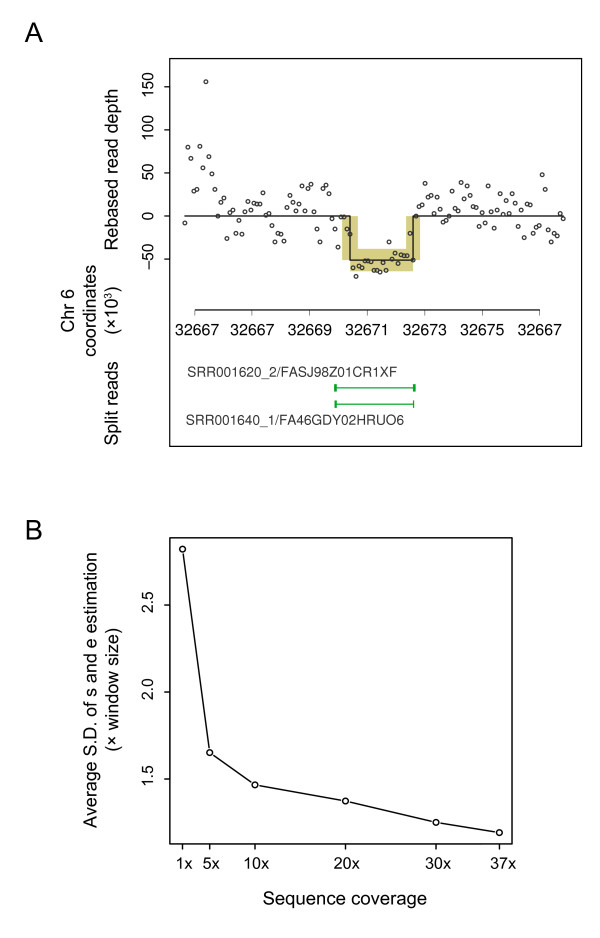
We developed a Bayesian statistical analysis algorithm for the detection of CNVs from both types of genomic data. The algorithm can analyze such data obtained from PCR-based bacterial artificial chromosome arrays, high-density oligonucleotide arrays, and more recently developed high-throughput DNA sequencing. Treating parameters--e.g., the number of CNVs, the position of each CNV, and the data noise level--that define the underlying data generating process as random variables, our approach derives the posterior distribution of the genomic CNV structure given the observed data. Sampling from the posterior distribution using a Markov chain Monte Carlo method, we get not only best estimates for these unknown parameters but also Bayesian credible intervals for the estimates. We illustrate the characteristics of our algorithm by applying it to both synthetic and experimental data sets in comparison to other segmentation algorithms.
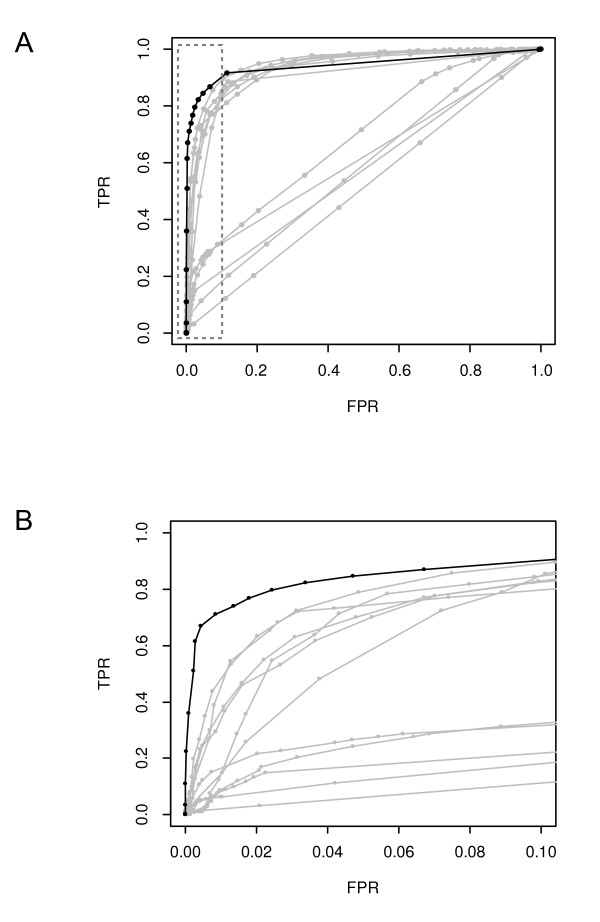
In particular, the synthetic data comparison shows that our method is more sensitive than other approaches at low false positive rates. Furthermore, given its Bayesian origin, our method can also be seen as a technique to refine CNVs identified by fast point-estimate methods and also as a framework to integrate array-CGH and sequencing data with other CNV-related biological knowledge, all through informative priors.
已证实拷贝数变异(CNVs)频繁发生,并被广泛认为对人类群体的表型变异有重要贡献。基于阵列的比较基因组杂交(array-CGH)和新开发的超高通量基因组测序的读深度方法都提供了快速、稳健和全面的方法,可在全基因组范围内识别 CNVs。
我们开发了一种贝叶斯统计分析算法,用于从这两种类型的基因组数据中检测 CNVs。该算法可以分析基于 PCR 的细菌人工染色体阵列、高密度寡核苷酸阵列以及最近开发的高通量 DNA 测序获得的数据。将定义潜在数据生成过程的参数(例如,CNV 的数量、每个 CNV 的位置以及数据噪声水平)视为随机变量,我们的方法可以从观察到的数据中推导出基因组 CNV 结构的后验分布。通过使用马尔可夫链蒙特卡罗方法从后验分布中进行抽样,我们不仅可以得到这些未知参数的最佳估计值,还可以得到估计值的贝叶斯可信区间。我们通过将其应用于合成数据集和实验数据集并与其他分割算法进行比较,展示了我们算法的特点。
特别是,与其他方法相比,合成数据的比较表明我们的方法在低假阳性率下更敏感。此外,由于其贝叶斯起源,我们的方法还可以被视为一种通过快速点估计方法来改进 CNV 的技术,也可以作为一种框架,通过信息丰富的先验知识将 array-CGH 和测序数据与其他与 CNV 相关的生物学知识整合在一起。