Department of Neurology, University of Texas Medical Branch, Galveston, Texas, USA.
J Neurosci Res. 2012 Mar;90(3):706-14. doi: 10.1002/jnr.22786. Epub 2011 Nov 8.
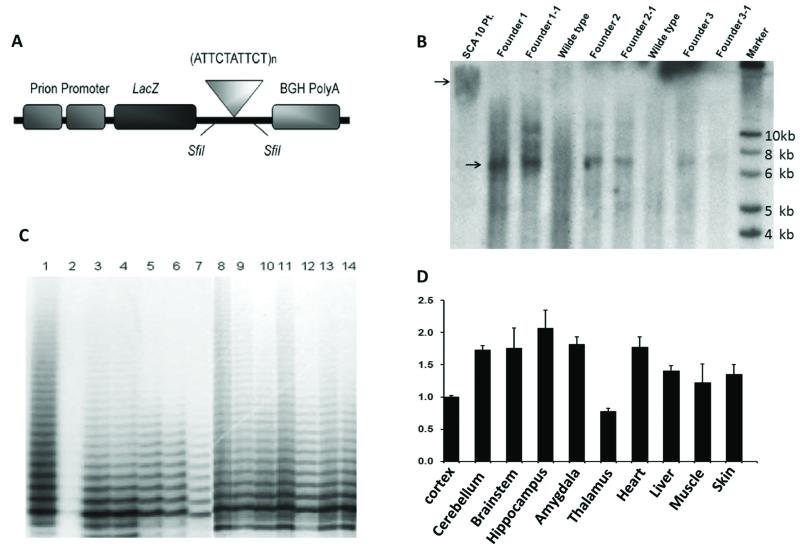
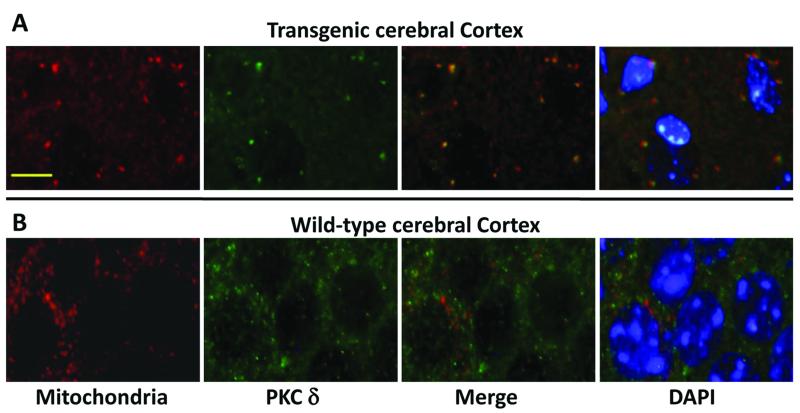
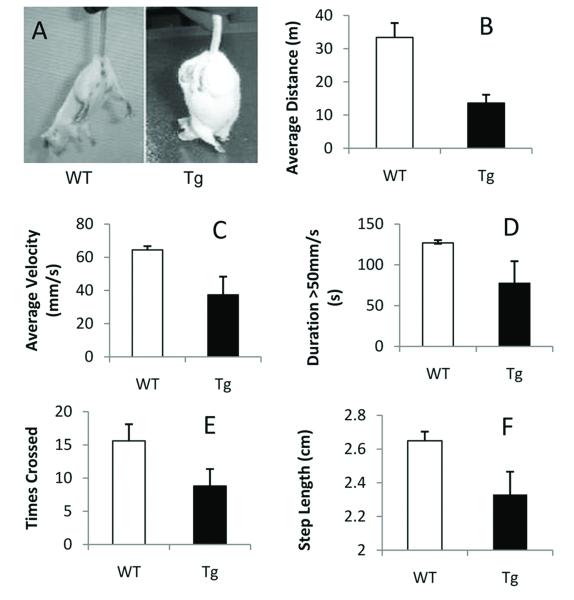
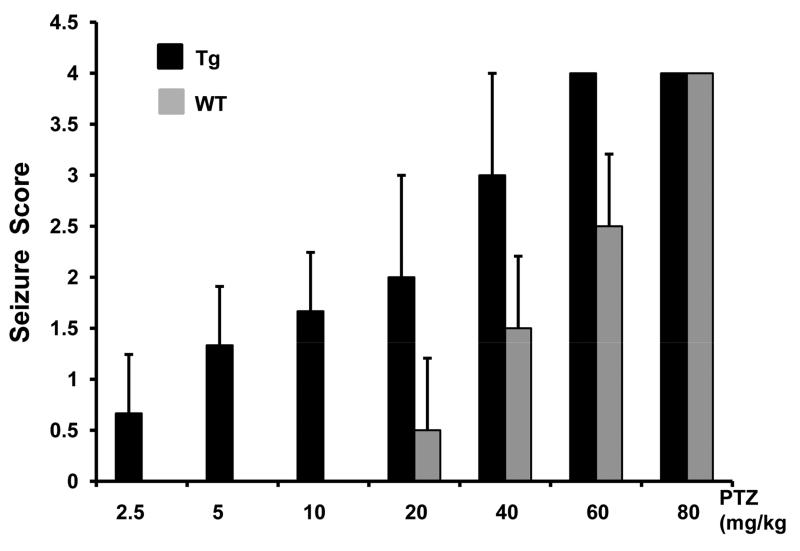
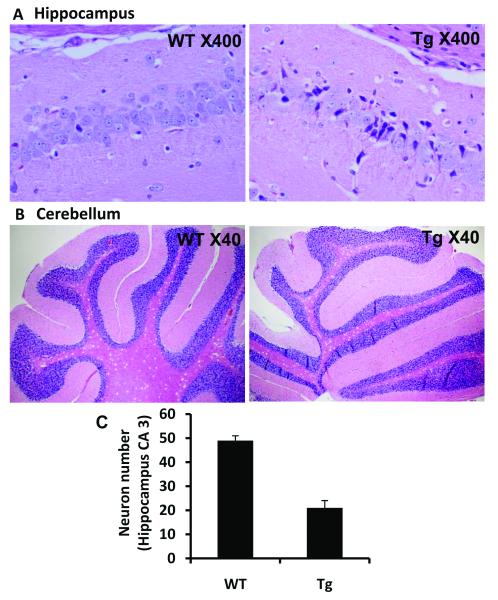
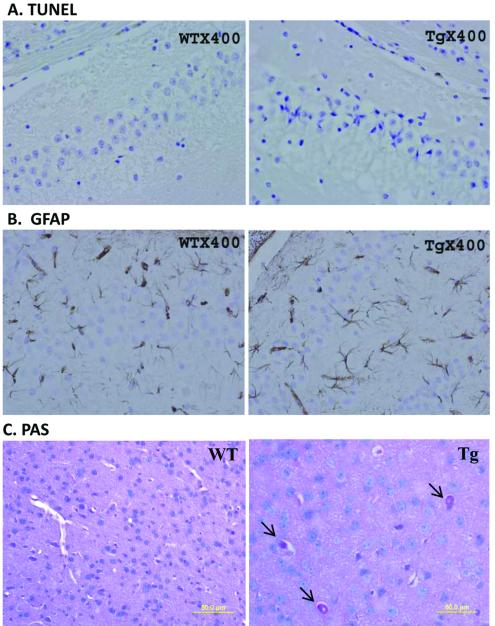
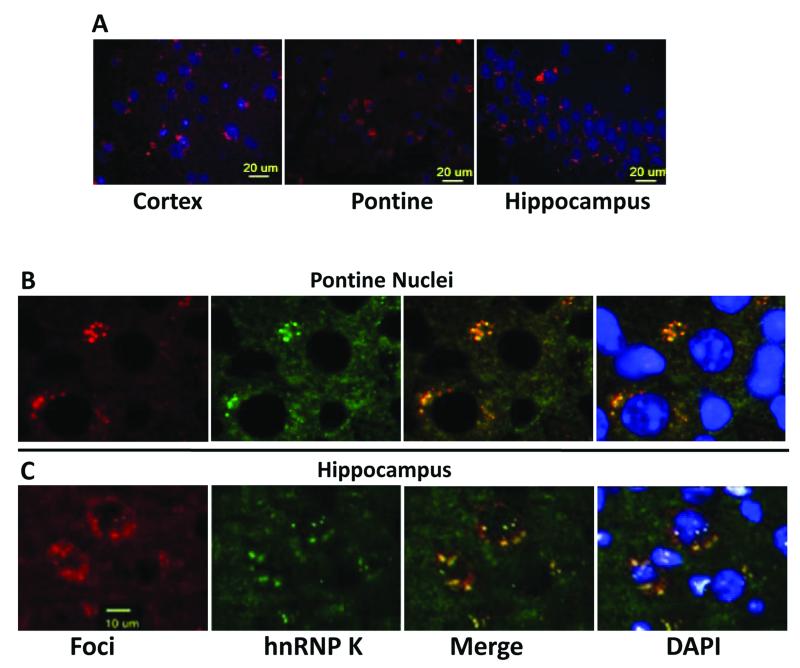
Spinocerebellar ataxia type 10 (SCA10) is an autosomal dominant neurodegenerative disorder manifested by ataxia and seizure. SCA10 is caused by a large expansion of an intronic ATTCT pentanucleotide repeat in the ATXN10 gene. We have recently postulated a toxic RNA-mediated gain of function in the pathogenesis of spinal cerebellar ataxia type 10 (SCA10). The spliced intron-9 RNA containing the expanded AUUCU repeat aggregates in SCA10 cells and sequesters hnRNP K. hnRNP K sequestration triggers the translocation of protein kinase Cδ (PKCδ) to mitochondria, leading to activation of caspase-3 and apoptosis. To confirm the toxic RNA-mediated gain of function, we generated a new transgenic mouse model in which the expanded pentanucleotide repeats are constructed in the 3'-untranslated region (3'UTR) to ensure transcription without translation of the repeat. We constructed an artificial transgene containing the SCA10 (ATTCT)(500) track within the 3'UTR of the LacZ gene driven by the rat prion promoter (PrP) and used this to generate a new transgenic mouse model for SCA10. We then examined these mice for neurological phenotypes and histopathological, molecular, and cellular changes. The transgenic mice showed irregular gait and increased seizure susceptibility at the age of 6 months, resembling the clinical phenotype of SCA10. The cerebral cortex, hippocampus, and pontine nuclei showed neuronal loss. The brains of these animals also showed molecular and cellular changes similar to those previously found in an SCA10 cell model. Expression of the expanded SCA10 AUUCU repeat within the 3'UTR of a gene results in neuronal loss with associated gait abnormalities and increased seizure susceptibility phenotypes, which resemble those seen in SCA10 patients. Moreover, these results bolster the idea that the SCA10 disease mechanism is mediated by a toxic RNA gain-of-function mutation of the expanded AUUCU repeat.
脊髓小脑共济失调 10 型(SCA10)是一种常染色体显性遗传性神经退行性疾病,表现为共济失调和癫痫。SCA10 是由 ATXN10 基因内含子中的 ATTCT 五核苷酸重复序列的大量扩增引起的。我们最近提出了一种毒性 RNA 介导的功能获得在脊髓小脑共济失调 10 型(SCA10)发病机制中的作用。含有扩增 AUUCU 重复的剪接内含子 9 RNA 在 SCA10 细胞中聚集,并隔离 hnRNP K。hnRNP K 的隔离触发蛋白激酶 Cδ(PKCδ)向线粒体易位,导致 caspase-3 的激活和细胞凋亡。为了证实毒性 RNA 介导的功能获得,我们构建了一种新的转基因小鼠模型,其中扩增的五核苷酸重复序列构建在 3'非翻译区(3'UTR)中,以确保重复序列的转录而不翻译。我们构建了一个包含 SCA10(ATTCT)(500)轨迹的人工转基因,该轨迹位于 LacZ 基因的 3'UTR 内,由大鼠朊病毒启动子(PrP)驱动,并使用该转基因构建了一种新的 SCA10 转基因小鼠模型。然后,我们检查了这些小鼠的神经表型以及组织病理学、分子和细胞变化。转基因小鼠在 6 个月时表现出不规则的步态和增加的癫痫易感性,类似于 SCA10 的临床表型。大脑皮层、海马和脑桥核显示神经元丢失。这些动物的大脑还显示出与以前在 SCA10 细胞模型中发现的类似的分子和细胞变化。在基因的 3'UTR 内表达扩增的 SCA10 AUUCU 重复导致神经元丢失,伴有相关的步态异常和增加的癫痫易感性表型,类似于 SCA10 患者的表型。此外,这些结果支持了 SCA10 疾病机制是由扩增的 AUUCU 重复的毒性 RNA 获得性功能突变介导的观点。