Centre for Genomic Regulation (CRG), Barcelona, Spain.
Genome Biol. 2011 Nov 8;12(11):R112. doi: 10.1186/gb-2011-12-11-r112.
The generation and analysis of high-throughput sequencing data are becoming a major component of many studies in molecular biology and medical research. Illumina's Genome Analyzer (GA) and HiSeq instruments are currently the most widely used sequencing devices. Here, we comprehensively evaluate properties of genomic HiSeq and GAIIx data derived from two plant genomes and one virus, with read lengths of 95 to 150 bases.
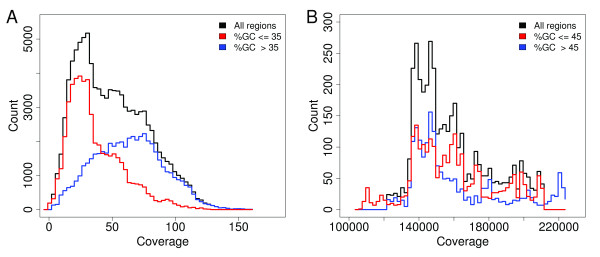
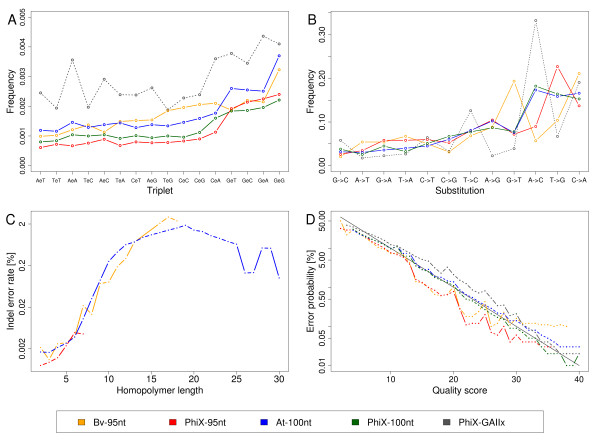
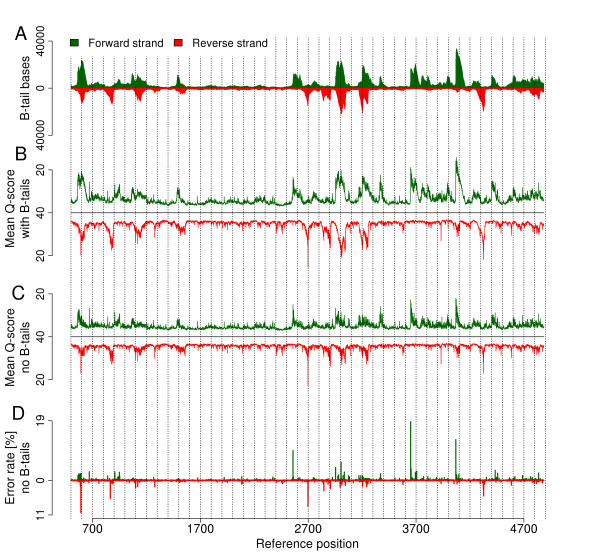
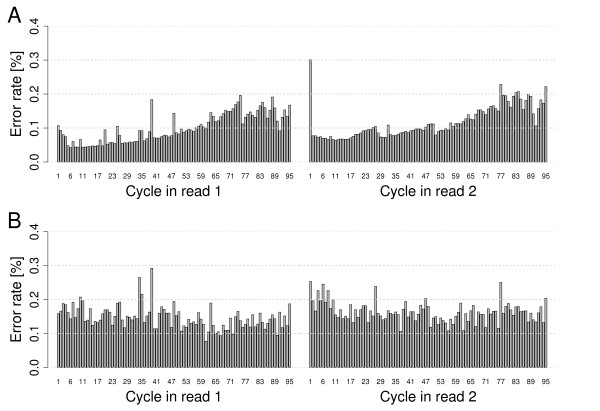
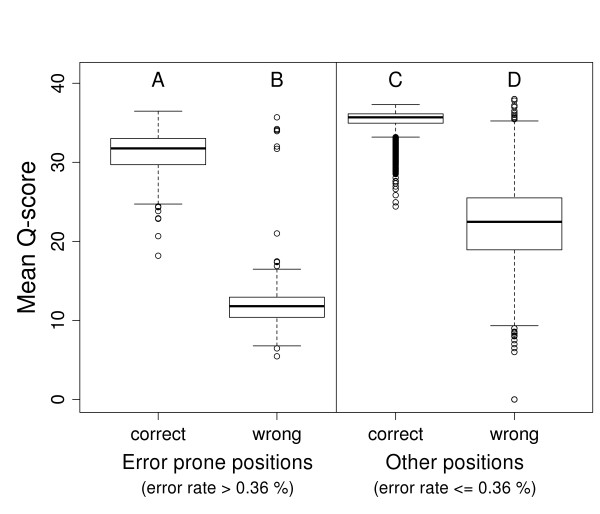
We provide quantifications and evidence for GC bias, error rates, error sequence context, effects of quality filtering, and the reliability of quality values. By combining different filtering criteria we reduced error rates 7-fold at the expense of discarding 12.5% of alignable bases. While overall error rates are low in HiSeq data we observed regions of accumulated wrong base calls. Only 3% of all error positions accounted for 24.7% of all substitution errors. Analyzing the forward and reverse strands separately revealed error rates of up to 18.7%. Insertions and deletions occurred at very low rates on average but increased to up to 2% in homopolymers. A positive correlation between read coverage and GC content was found depending on the GC content range.
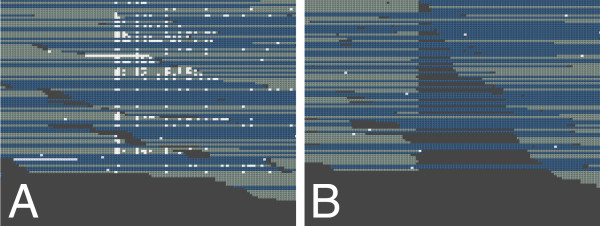
The errors and biases we report have implications for the use and the interpretation of Illumina sequencing data. GAIIx and HiSeq data sets show slightly different error profiles. Quality filtering is essential to minimize downstream analysis artifacts. Supporting previous recommendations, the strand-specificity provides a criterion to distinguish sequencing errors from low abundance polymorphisms.
高通量测序数据的产生和分析正成为分子生物学和医学研究中许多研究的主要组成部分。Illumina 的 Genome Analyzer(GA)和 HiSeq 仪器是目前使用最广泛的测序设备。在这里,我们全面评估了两个植物基因组和一个病毒的基因组 HiSeq 和 GAIIx 数据的特性,读取长度为 95 到 150 个碱基。
我们提供了 GC 偏倚、错误率、错误序列上下文、质量过滤的影响以及质量值的可靠性的定量和证据。通过结合不同的过滤标准,我们将错误率降低了 7 倍,但代价是丢弃了 12.5%的可对齐碱基。虽然 HiSeq 数据中的总体错误率较低,但我们观察到了累积错误碱基调用的区域。所有错误位置仅占所有取代错误的 24.7%,而占所有错误位置的 3%。分别分析正向和反向链,发现错误率高达 18.7%。平均而言,插入和缺失的发生率非常低,但在同聚体中增加到 2%。发现读取覆盖率与 GC 含量之间存在正相关关系,具体取决于 GC 含量范围。
我们报告的错误和偏差对 Illumina 测序数据的使用和解释有影响。GAIIx 和 HiSeq 数据集显示出略有不同的错误分布。质量过滤对于最小化下游分析伪影至关重要。支持先前的建议,链特异性提供了区分测序错误和低丰度多态性的标准。