Schirmer Melanie, D'Amore Rosalinda, Ijaz Umer Z, Hall Neil, Quince Christopher
The Broad Institute of MIT and Harvard, 415 Main Street, Cambridge, MA 02142, USA.
Harvard T.H. Chan School of Public Health, 655 Huntington Ave, Boston, MA 02115, USA.
BMC Bioinformatics. 2016 Mar 11;17:125. doi: 10.1186/s12859-016-0976-y.
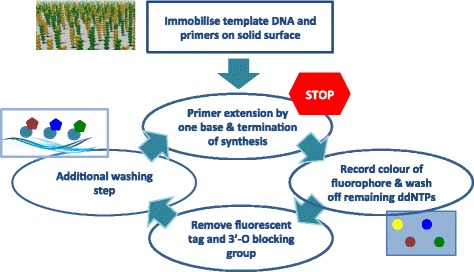
Illumina's sequencing platforms are currently the most utilised sequencing systems worldwide. The technology has rapidly evolved over recent years and provides high throughput at low costs with increasing read-lengths and true paired-end reads. However, data from any sequencing technology contains noise and our understanding of the peculiarities and sequencing errors encountered in Illumina data has lagged behind this rapid development.
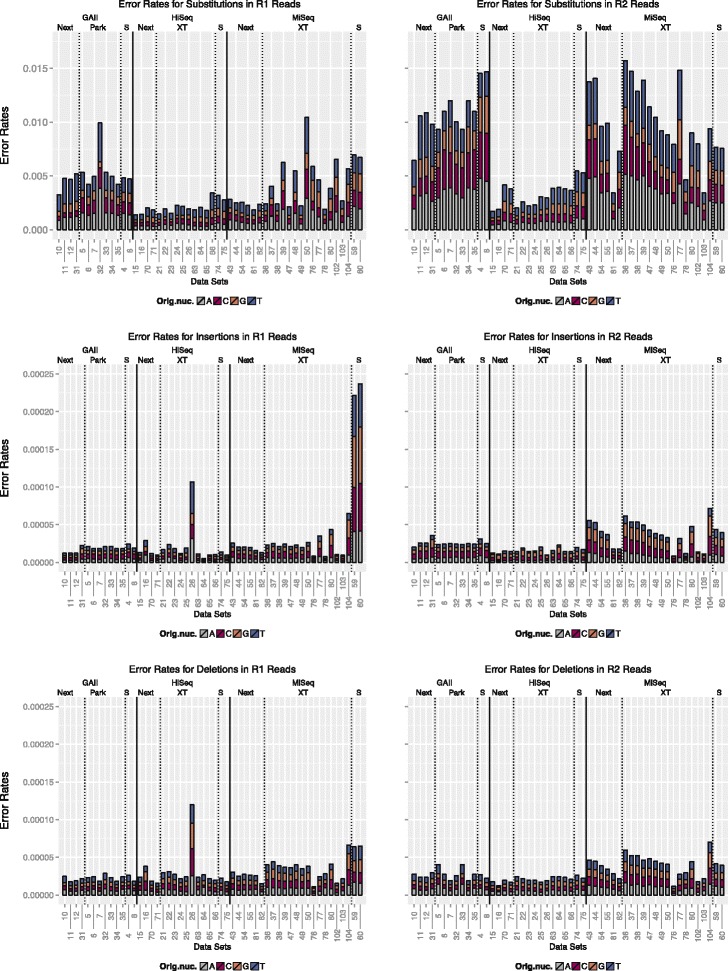
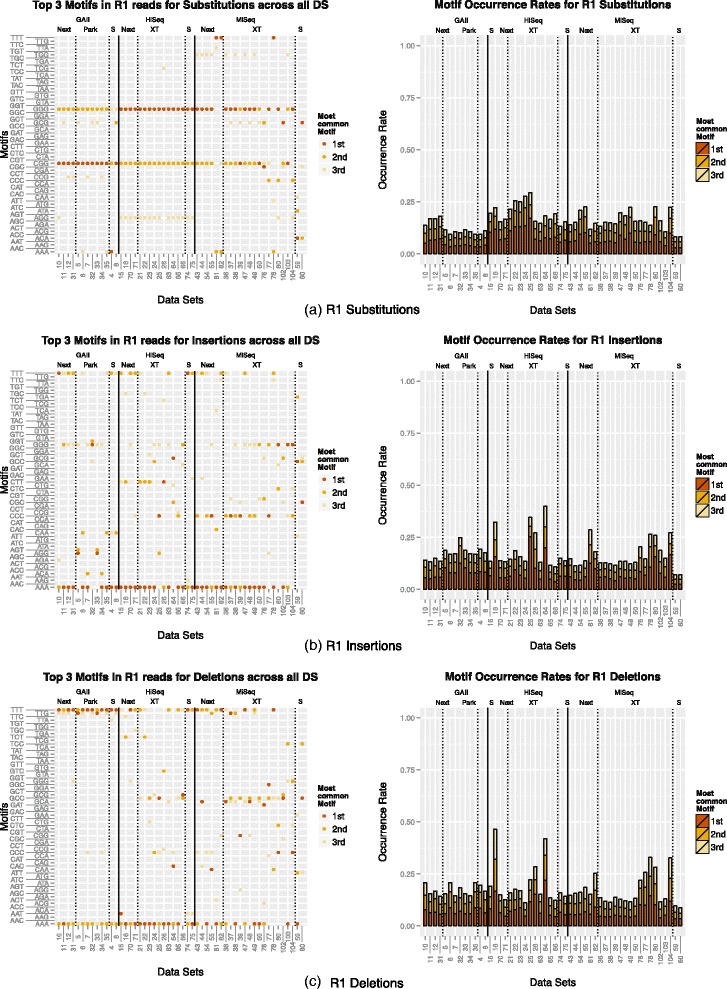
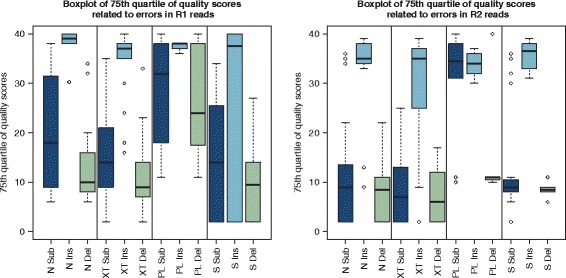
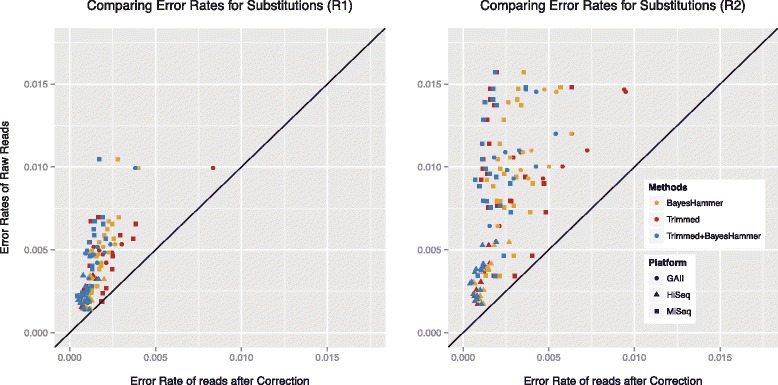
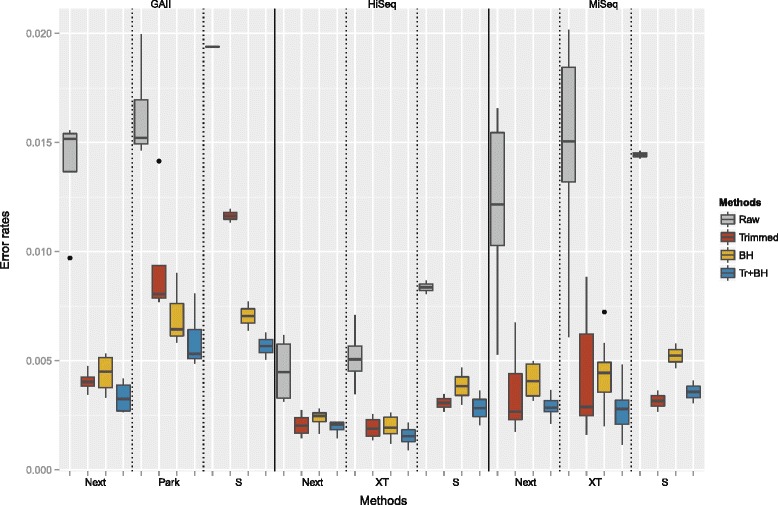
We conducted a systematic investigation of errors and biases in Illumina data based on the largest collection of in vitro metagenomic data sets to date. We evaluated the Genome Analyzer II, HiSeq and MiSeq and tested state-of-the-art low input library preparation methods. Analysing in vitro metagenomic sequencing data allowed us to determine biases directly associated with the actual sequencing process. The position- and nucleotide-specific analysis revealed a substantial bias related to motifs (3mers preceding errors) ending in "GG". On average the top three motifs were linked to 16 % of all substitution errors. Furthermore, a preferential incorporation of ddGTPs was recorded. We hypothesise that all of these biases are related to the engineered polymerase and ddNTPs which are intrinsic to any sequencing-by-synthesis method. We show that quality-score-based error removal strategies can on average remove 69 % of the substitution errors - however, the motif-bias remains.
Single-nucleotide polymorphism changes in bacterial genomes can cause significant changes in phenotype, including antibiotic resistance and virulence, detecting them within metagenomes is therefore vital. Current error removal techniques are not designed to target the peculiarities encountered in Illumina sequencing data and other sequencing-by-synthesis methods, causing biases to persist and potentially affect any conclusions drawn from the data. In order to develop effective diagnostic and therapeutic approaches we need to be able to identify systematic sequencing errors and distinguish these errors from true genetic variation.
Illumina的测序平台是目前全球使用最广泛的测序系统。近年来,该技术迅速发展,能够以低成本实现高通量测序,同时读长不断增加,真正的双端测序也得以实现。然而,任何测序技术产生的数据都包含噪声,我们对Illumina数据中出现的特性和测序错误的理解,却落后于这一快速发展的技术。
我们基于迄今为止最大规模的体外宏基因组数据集,对Illumina数据中的错误和偏差进行了系统研究。我们评估了Genome Analyzer II、HiSeq和MiSeq,并测试了最先进的低输入文库制备方法。分析体外宏基因组测序数据使我们能够直接确定与实际测序过程相关的偏差。位置和核苷酸特异性分析揭示了与以“GG”结尾的基序(错误前的三联体)相关的显著偏差。平均而言,排名前三的基序与所有替换错误的16%相关。此外,还记录到ddGTP的优先掺入。我们推测所有这些偏差都与工程化聚合酶和ddNTPs有关,而它们是任何合成测序方法所固有的。我们表明,基于质量评分的错误去除策略平均可以去除69%的替换错误——然而,基序偏差仍然存在。
细菌基因组中的单核苷酸多态性变化可导致表型的显著变化,包括抗生素抗性和毒力,因此在宏基因组中检测这些变化至关重要。当前的错误去除技术并非针对Illumina测序数据及其他合成测序方法中出现的特性而设计,导致偏差持续存在,并可能影响从数据中得出的任何结论。为了开发有效的诊断和治疗方法我们需要能够识别系统的测序错误,并将这些错误与真正的基因变异区分开来。