McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA.
Nat Rev Genet. 2011 Nov 29;13(1):36-46. doi: 10.1038/nrg3117.
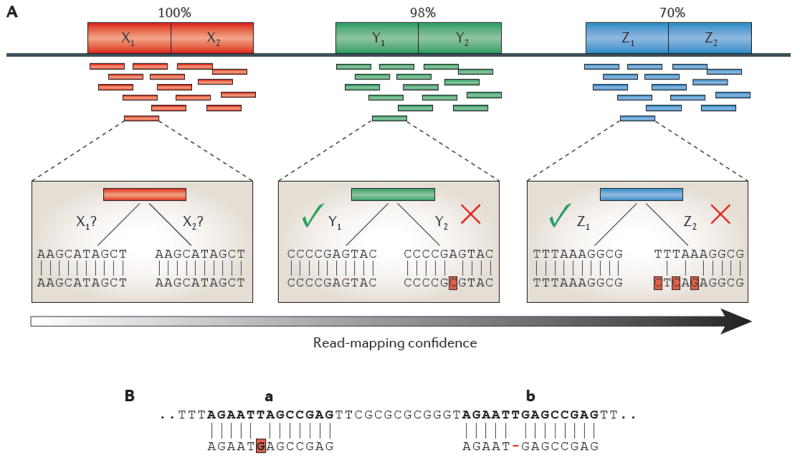
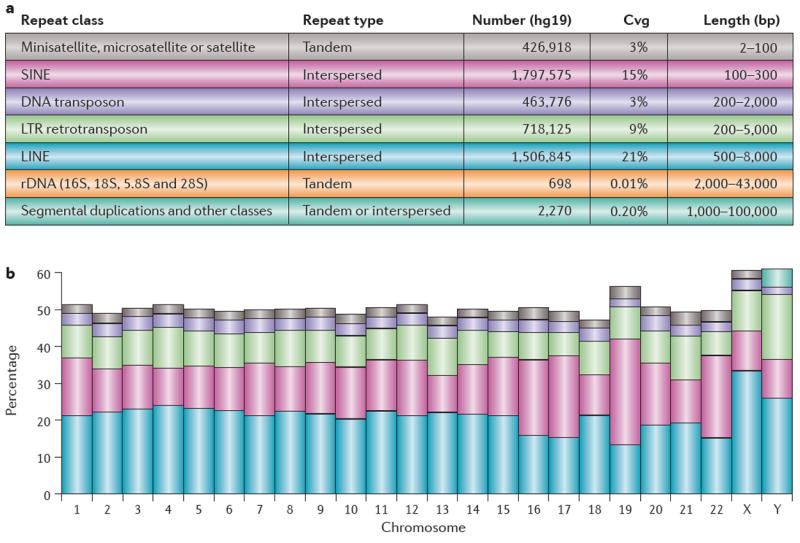
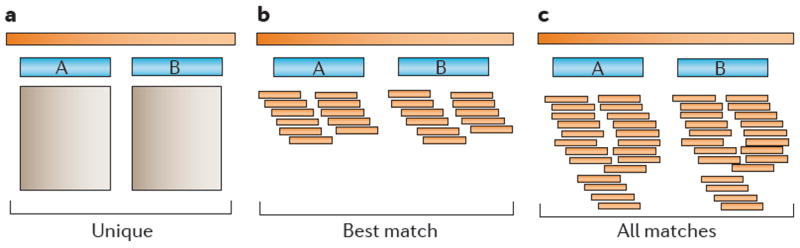
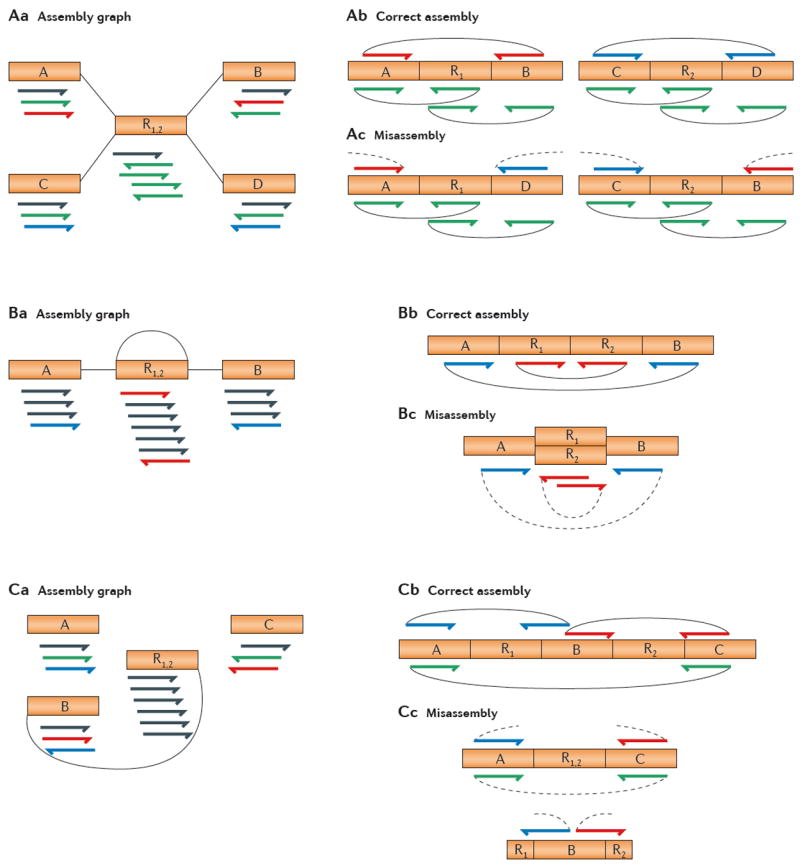
Repetitive DNA sequences are abundant in a broad range of species, from bacteria to mammals, and they cover nearly half of the human genome. Repeats have always presented technical challenges for sequence alignment and assembly programs. Next-generation sequencing projects, with their short read lengths and high data volumes, have made these challenges more difficult. From a computational perspective, repeats create ambiguities in alignment and assembly, which, in turn, can produce biases and errors when interpreting results. Simply ignoring repeats is not an option, as this creates problems of its own and may mean that important biological phenomena are missed. We discuss the computational problems surrounding repeats and describe strategies used by current bioinformatics systems to solve them.
重复 DNA 序列在从细菌到哺乳动物的广泛物种中都很丰富,它们覆盖了人类基因组的近一半。重复序列一直是序列比对和组装程序的技术挑战。具有短读长和大数据量的新一代测序项目使这些挑战更加困难。从计算的角度来看,重复序列在比对和组装中造成了不确定性,而这反过来又会在解释结果时产生偏差和错误。简单地忽略重复序列不是一个可行的选择,因为这会产生自身的问题,并且可能意味着重要的生物学现象被遗漏。我们讨论了围绕重复序列的计算问题,并描述了当前生物信息学系统用来解决这些问题的策略。