US Military HIV Research Program (MHRP), Rockville, MD, USA.
J Transl Med. 2011 Dec 8;9:212. doi: 10.1186/1479-5876-9-212.
HIV vaccine development must address the genetic diversity and plasticity of the virus that permits the presentation of diverse genetic forms to the immune system and subsequent escape from immune pressure. Assessment of potential HIV strain coverage by candidate T cell-based vaccines (whether natural sequence or computationally optimized products) is now a critical component in interpreting candidate vaccine suitability.
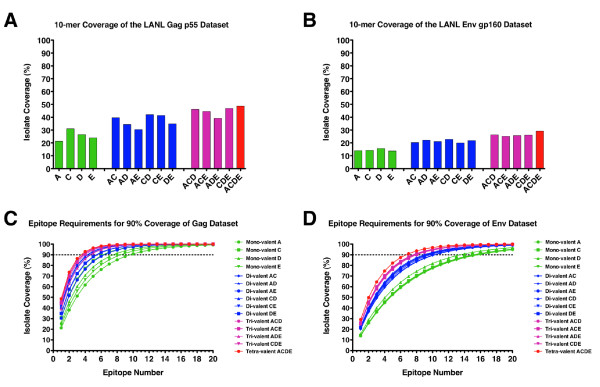
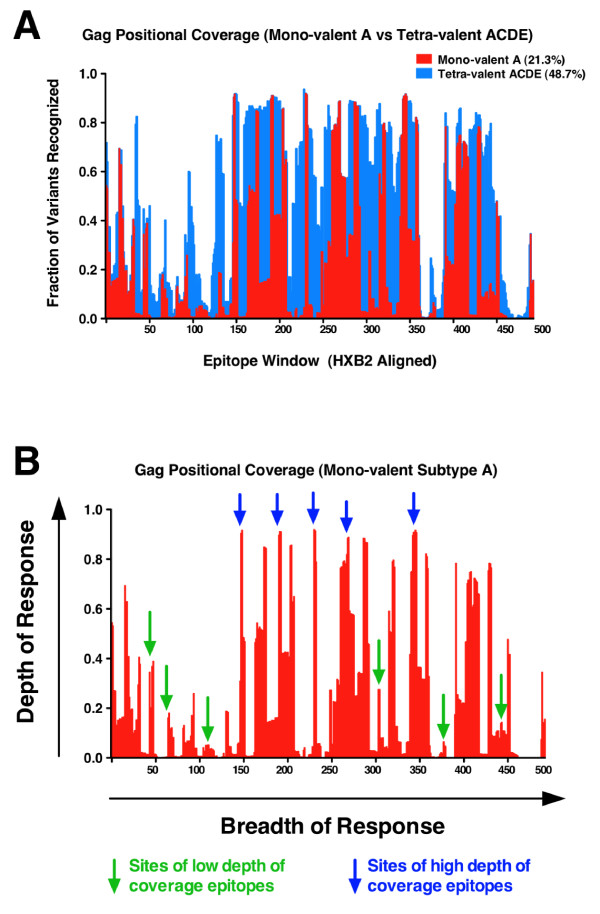
We have utilized an N-mer identity algorithm to represent T cell epitopes and explore potential coverage of the global HIV pandemic using natural sequences derived from candidate HIV vaccines. Breadth (the number of T cell epitopes generated) and depth (the variant coverage within a T cell epitope) analyses have been incorporated into the model to explore vaccine coverage requirements in terms of the number of discrete T cell epitopes generated.
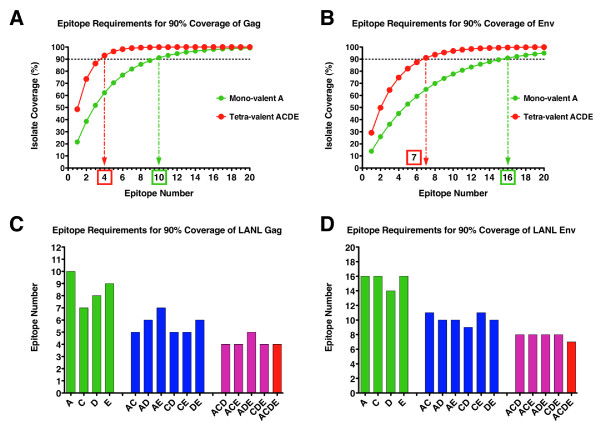
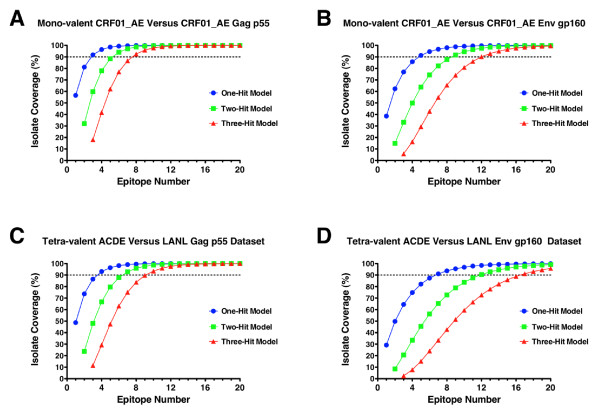
We show that when multiple epitope generation by a vaccine product is considered a far more nuanced appraisal of the potential HIV strain coverage of the vaccine product emerges. By considering epitope breadth and depth several important observations were made: (1) epitope breadth requirements to reach particular levels of vaccine coverage, even for natural sequence-based vaccine products is not necessarily an intractable problem for the immune system; (2) increasing the valency (number of T cell epitope variants present) of vaccine products dramatically decreases the epitope requirements to reach particular coverage levels for any epidemic; (3) considering multiple-hit models (more than one exact epitope match with an incoming HIV strain) places a significantly higher requirement upon epitope breadth in order to reach a given level of coverage, to the point where low valency natural sequence based products would not practically be able to generate sufficient epitopes.
When HIV vaccine sequences are compared against datasets of potential incoming viruses important metrics such as the minimum epitope count required to reach a desired level of coverage can be easily calculated. We propose that such analyses can be applied early in the planning stages and during the execution phase of a vaccine trial to explore theoretical and empirical suitability of a vaccine product to a particular epidemic setting.
HIV 疫苗的开发必须解决病毒的遗传多样性和可塑性问题,这使得病毒能够向免疫系统呈现多种遗传形式,并随后逃避免疫压力。评估候选 T 细胞疫苗(无论是天然序列还是经过计算优化的产品)对潜在 HIV 株的覆盖范围,现在是解释候选疫苗适用性的一个关键组成部分。
我们利用 N- 聚体同一性算法来表示 T 细胞表位,并利用来自候选 HIV 疫苗的天然序列探索全球 HIV 大流行的潜在覆盖范围。广度(生成的 T 细胞表位数量)和深度(T 细胞表位内的变体覆盖范围)分析已纳入模型中,以根据生成的离散 T 细胞表位数量探索疫苗覆盖范围要求。
我们表明,当考虑疫苗产品的多个表位生成时,疫苗产品对潜在 HIV 株的覆盖范围的评估会更加细致入微。通过考虑表位的广度和深度,我们得出了几个重要的观察结果:(1)即使对于基于天然序列的疫苗产品,达到特定疫苗覆盖水平所需的表位广度要求也不一定是免疫系统的难题;(2)增加疫苗产品的效价(存在的 T 细胞表位变体数量)会极大地降低达到任何流行水平特定覆盖水平所需的表位数量;(3)考虑多击模型(与传入的 HIV 株有多个精确的表位匹配)会对达到给定覆盖水平所需的表位广度提出更高的要求,以至于低效价的基于天然序列的产品实际上无法产生足够的表位。
当将 HIV 疫苗序列与潜在传入病毒的数据集进行比较时,可以轻松计算达到所需覆盖水平所需的最小表位数量等重要指标。我们提出,此类分析可以在疫苗试验的规划阶段早期和执行阶段进行,以探索疫苗产品对特定流行环境的理论和经验适用性。