Department of Medical Chemistry, Kansai Medical University, Moriguchi, Japan.
Mol Pain. 2011 Dec 22;7:101. doi: 10.1186/1744-8069-7-101.
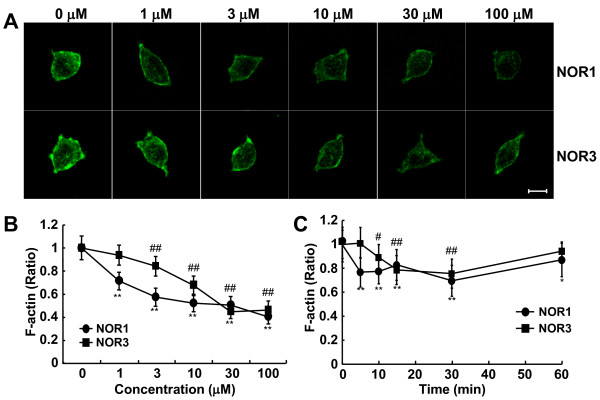
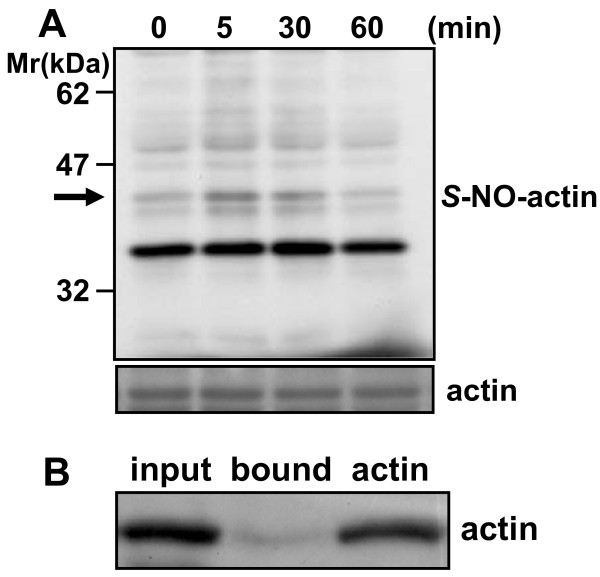
S-Nitrosylation, the reversible post-translational modification of reactive cysteine residues in proteins, has emerged as an important mechanism by which NO acts as a signaling molecule. We recently demonstrated that actin is a major S-nitrosylated protein in the spinal cord and suggested that NO directly attenuates dopamine release from PC12 cells by causing the breakdown of F-actin. However, the occurrence of S-nitrosylation of actin remained unclarified in animal pain model. Kinetic analysis of S-nitrosylation of actin in the present study was made by using NO-generating donors. The biotin-switch assay and purification on streptavidin-agarose were employed for identification of S-nitrosylated actin.
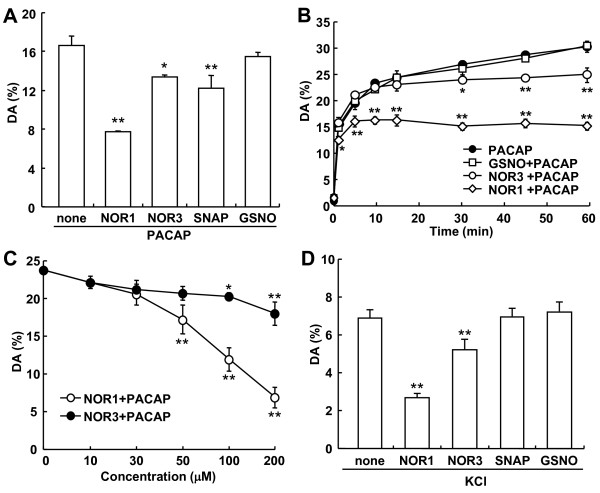
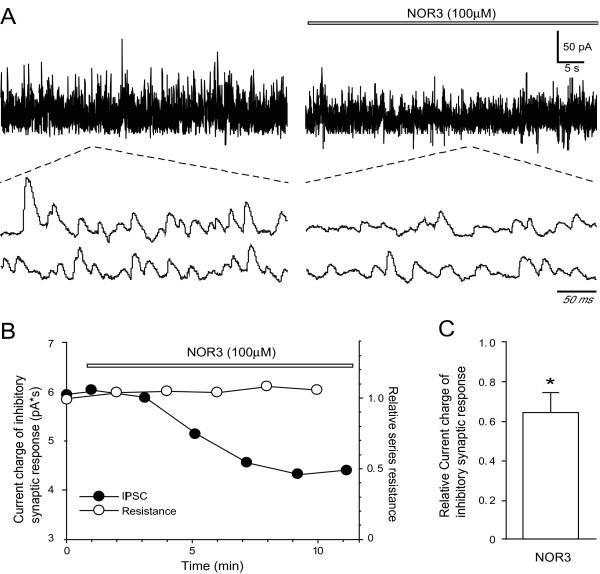
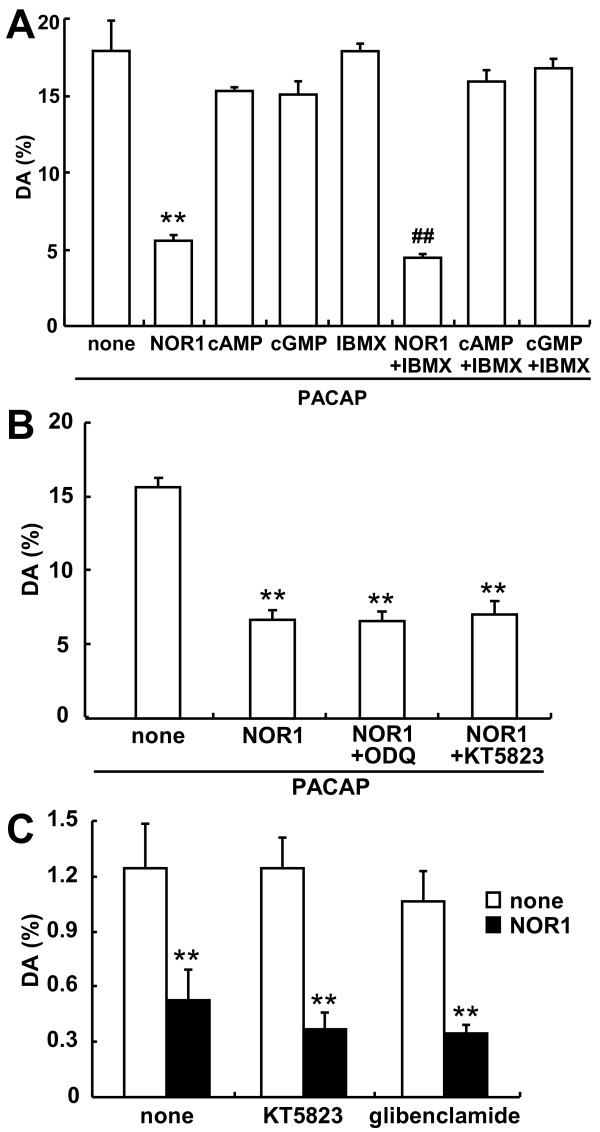
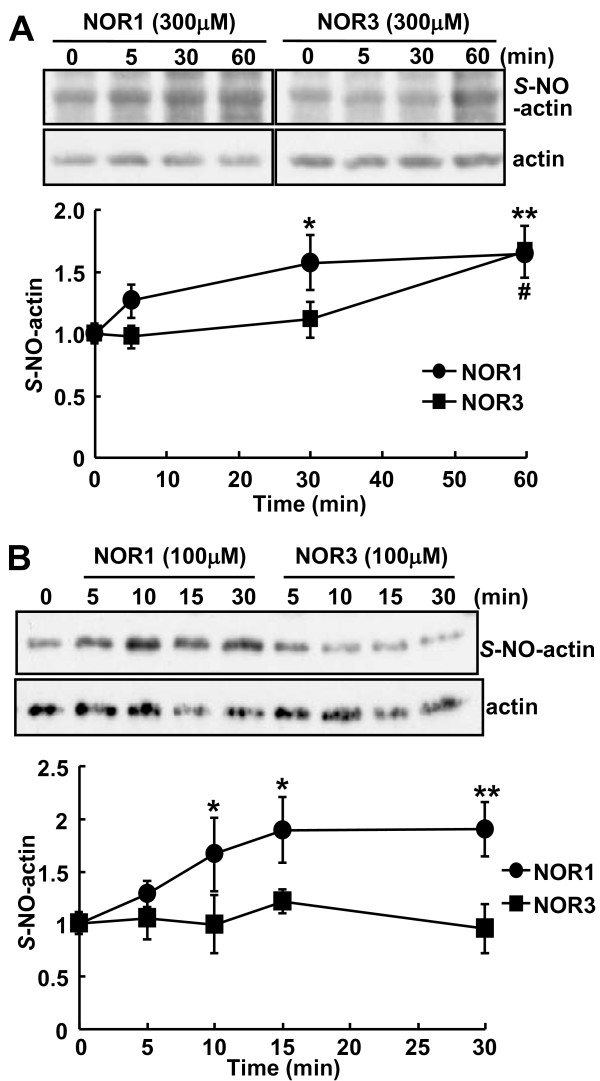
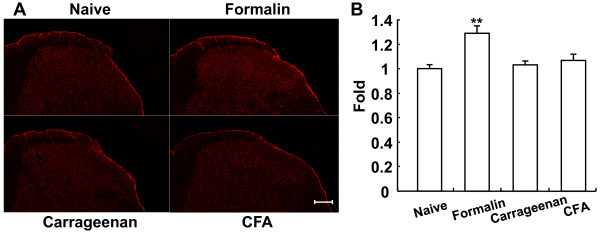
Dopamine release from PC12 cells was markedly attenuated by NOR1 (t1/2 = 1.8 min) and much less by NOR3 (t1/2 = 30 min), but not by S-nitroso-glutathione, an endogenous NO donor. A membrane-permeable cGMP analogue could not substitute for NOR1 as a suppressor nor could inhibitors of soluble guanylate cyclase and cGMP-dependent protein kinase attenuate the suppression. S-Nitrosylated actin was detected by the biotin-switch assay at 5 min after the addition of NOR1. Consistent with the kinetic analysis, actin in the spinal cord was rapidly and maximally S-nitrosylated in an inflammatory pain model at 5 min after the injection of 2% formalin into the hind paws. In vivo patch-clamp recordings of the spinal dorsal horn, NOR3 showed an inhibitory action on inhibitory synaptic transmission in interneurons of the substantia gelatinosa.
The present study demonstrates that rapid S-nitrosylation of actin occurred in vitro in the presence of exogenous NO-generating donors and in vivo in inflammatory pain model mice. Our data suggest that, in addition to the well-known cGMP-dependent protein kinase pathway, S-nitrosylation is involved in pain transmission via disinhibition of inhibitory neurons.
蛋白质中活性半胱氨酸残基的可逆翻译后修饰 S-亚硝基化作用,已成为 NO 作为信号分子发挥作用的重要机制。我们最近证明,肌动蛋白是脊髓中主要的 S-亚硝基化蛋白,并表明 NO 通过引起 F-肌动蛋白的分解,直接减弱 PC12 细胞中多巴胺的释放。然而,在动物疼痛模型中,肌动蛋白的 S-亚硝基化的发生仍不清楚。本研究通过使用 NO 生成供体对肌动蛋白 S-亚硝基化的动力学分析。采用生物素转移酶测定法和链霉亲和素琼脂糖纯化法鉴定 S-亚硝基化肌动蛋白。
NOR1(t1/2 = 1.8 分钟)明显减弱 PC12 细胞中多巴胺的释放,而 NOR3(t1/2 = 30 分钟)的作用较小,但 S-硝基谷胱甘肽,一种内源性 NO 供体,没有作用。膜通透型 cGMP 类似物不能替代 NOR1 作为抑制剂,可溶性鸟苷酸环化酶和 cGMP 依赖性蛋白激酶的抑制剂也不能减弱抑制作用。NOR1 添加 5 分钟后,通过生物素转移酶测定法检测到 S-亚硝基化肌动蛋白。与动力学分析一致,在向足底注射 2%甲醛后 5 分钟,炎症性疼痛模型中,脊髓中的肌动蛋白迅速且最大程度地被 S-亚硝基化。在脊髓背角的体内膜片钳记录中,NOR3 对胶质层中间神经元的抑制性突触传递显示出抑制作用。
本研究表明,在外源性 NO 生成供体存在的情况下,体外快速发生肌动蛋白 S-亚硝基化,体内在炎症性疼痛模型小鼠中也发生 S-亚硝基化。我们的数据表明,除了众所周知的 cGMP 依赖性蛋白激酶途径外,S-亚硝基化还通过抑制性神经元的去抑制参与疼痛传递。