Department of Pharmacology and Toxicology, Dartmouth Medical School, Lebanon, New Hampshire, USA.
Mol Cancer Ther. 2012 Feb;11(2):427-38. doi: 10.1158/1535-7163.MCT-11-0406. Epub 2011 Dec 27.
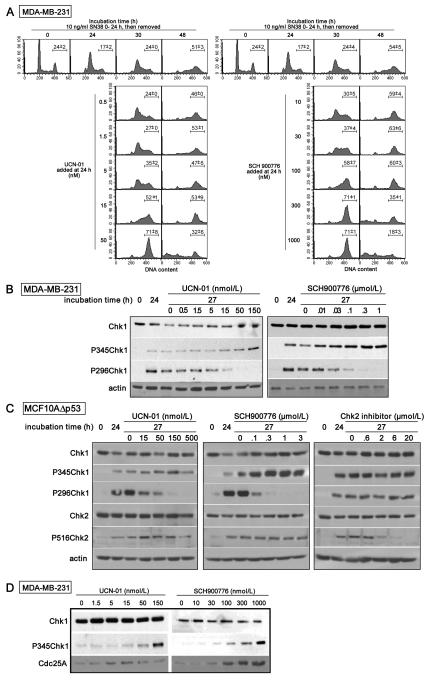
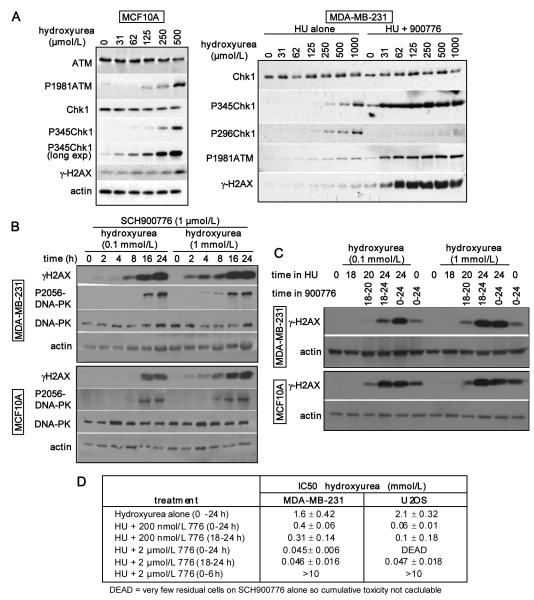
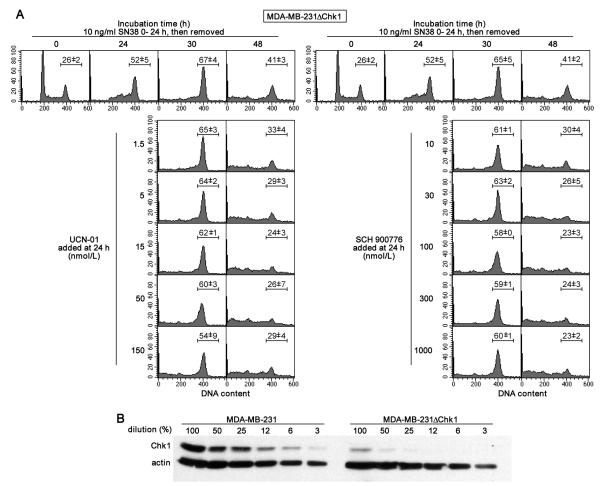
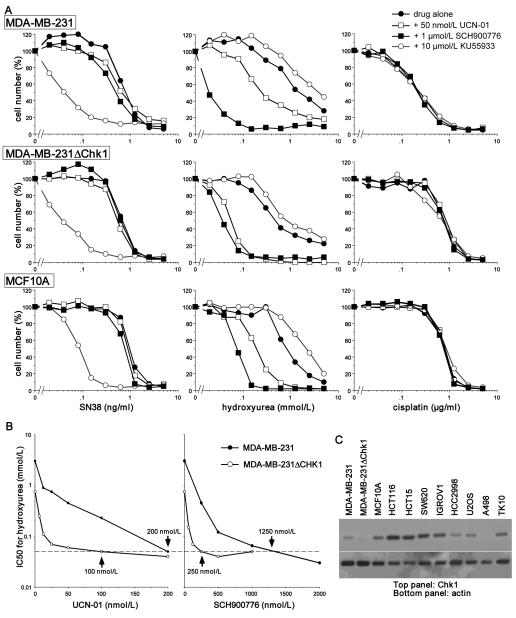
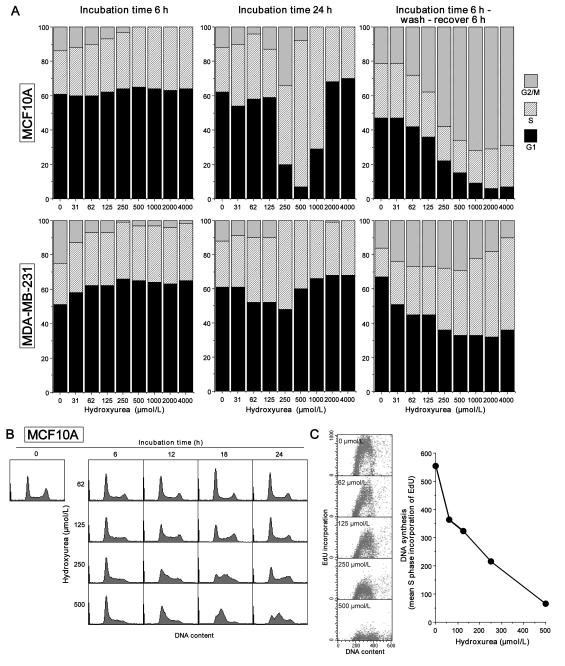
Many anticancer agents damage DNA and arrest cell-cycle progression primarily in S or G(2) phase of the cell cycle. Previous studies with the topoisomerase I inhibitor SN38 have shown the efficacy of the Chk1 inhibitor UCN-01 to overcome this arrest and induce mitotic catastrophe. UCN-01 was limited in clinical trials by unfavorable pharmacokinetics. SCH900776 is a novel and more selective Chk1 inhibitor that potently inhibits Chk1 and abrogates cell-cycle arrest induced by SN38. Like UCN-01, abrogation of SN38-induced arrest enhances the rate of cell death but does not increase overall cell death. In contrast, SCH900776 reduced the growth-inhibitory concentration of hydroxyurea by 20- to 70-fold. A similar magnitude of sensitization was observed with cytarabine. A 5- to 10-fold sensitization occurred with gemcitabine, but no sensitization occurred with cisplatin, 5-fluorouracil, or 6-thioguanine. Sensitization occurred at hydroxyurea concentrations that marginally slowed DNA replication without apparent activation of Chk1, but this led to dependence on Chk1 that increased with time. For example, when added 18 hours after hydroxyurea, SCH900776 induced DNA double-strand breaks consistent with rapid collapse of replication forks. In addition, some cell lines were highly sensitive to SCH900776 alone, and these cells required lower concentrations of SCH900776 to sensitize them to hydroxyurea. We conclude that some tumors may be very sensitive to the combination of SCH900776 and hydroxyurea. Delayed administration of SCH900776 may be more effective than concurrent treatment. SCH900776 is currently in phase I clinical trials, and these results provide the rationale and schedule for future clinical trials.
许多抗癌药物主要通过破坏 DNA 和阻滞细胞周期进程来发挥作用,其阻滞作用发生于细胞周期的 S 期或 G2 期。先前使用拓扑异构酶 I 抑制剂 SN38 的研究表明,细胞周期检验点激酶 1(Chk1)抑制剂 UCN-01 可以克服这种阻滞作用,诱导有丝分裂灾难。但 UCN-01 在临床试验中受到了不理想的药代动力学的限制。SCH900776 是一种新型的、更具选择性的 Chk1 抑制剂,它可以有效地抑制 Chk1,并消除 SN38 诱导的细胞周期阻滞。与 UCN-01 一样,消除 SN38 诱导的阻滞作用会增加细胞死亡的速度,但不会增加总体细胞死亡。相反,SCH900776 将羟基脲的生长抑制浓度降低了 20 至 70 倍。用阿糖胞苷也观察到了类似程度的增敏作用。用吉西他滨时增敏 5 至 10 倍,但用顺铂、5-氟尿嘧啶或 6-硫鸟嘌呤时则没有增敏作用。在羟脲浓度下,这种增敏作用轻微地减缓了 DNA 复制,但没有明显激活 Chk1,导致对 Chk1 的依赖性随着时间的推移而增加。例如,当在羟脲加入 18 小时后,SCH900776 诱导了 DNA 双链断裂,这与复制叉的迅速崩溃一致。此外,一些细胞系对 SCH900776 本身非常敏感,并且这些细胞需要更低浓度的 SCH900776 来使它们对羟脲增敏。我们得出结论,一些肿瘤可能对 SCH900776 和羟脲的组合非常敏感。延迟给予 SCH900776 可能比同时治疗更有效。SCH900776 目前正在进行 I 期临床试验,这些结果为未来的临床试验提供了依据和时间表。