The Human Genome Sequencing Center, Baylor College of Medicine, Houston, USA.
BMC Bioinformatics. 2012 Jan 12;13:8. doi: 10.1186/1471-2105-13-8.
Whole exome capture sequencing allows researchers to cost-effectively sequence the coding regions of the genome. Although the exome capture sequencing methods have become routine and well established, there is currently a lack of tools specialized for variant calling in this type of data.
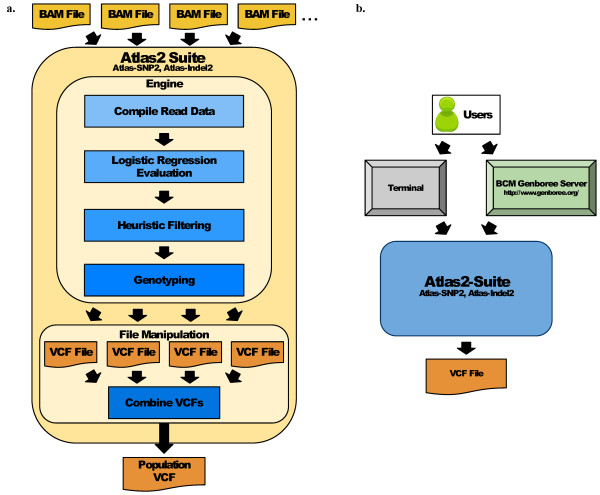
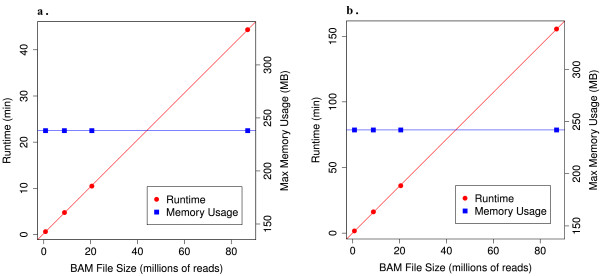
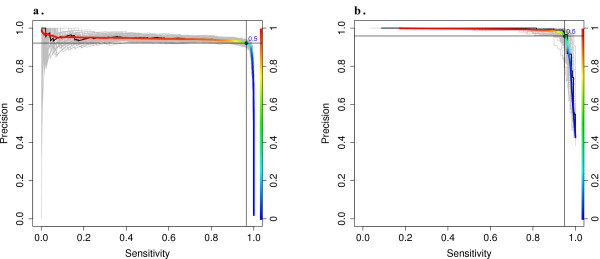
Using statistical models trained on validated whole-exome capture sequencing data, the Atlas2 Suite is an integrative variant analysis pipeline optimized for variant discovery on all three of the widely used next generation sequencing platforms (SOLiD, Illumina, and Roche 454). The suite employs logistic regression models in conjunction with user-adjustable cutoffs to accurately separate true SNPs and INDELs from sequencing and mapping errors with high sensitivity (96.7%).
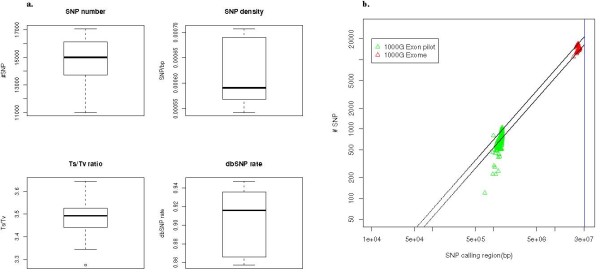
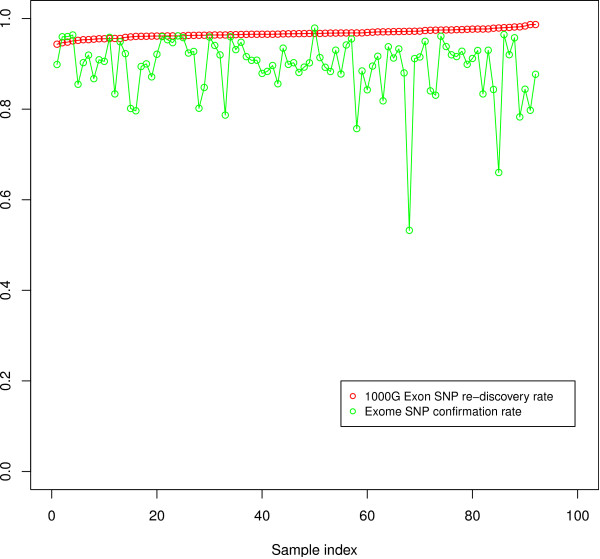
We have implemented the Atlas2 Suite and applied it to 92 whole exome samples from the 1000 Genomes Project. The Atlas2 Suite is available for download at http://sourceforge.net/projects/atlas2/. In addition to a command line version, the suite has been integrated into the Genboree Workbench, allowing biomedical scientists with minimal informatics expertise to remotely call, view, and further analyze variants through a simple web interface. The existing genomic databases displayed via the Genboree browser also streamline the process from variant discovery to functional genomics analysis, resulting in an off-the-shelf toolkit for the broader community.
全外显子组捕获测序可让研究人员经济有效地对基因组的编码区域进行测序。尽管外显子组捕获测序方法已变得常规且成熟,但目前缺乏专门针对此类数据进行变异调用的工具。
利用基于经验证的全外显子捕获测序数据训练的统计模型,Atlas2 套件是一种整合的变异分析管道,针对三种广泛使用的下一代测序平台(SOLiD、Illumina 和 Roche 454)进行了优化,可用于发现变异。该套件采用逻辑回归模型,并结合用户可调整的截断值,以高灵敏度(96.7%)准确地将真正的 SNP 和 INDEL 与测序和映射错误区分开来。
我们已经实现了 Atlas2 套件,并将其应用于 1000 基因组计划的 92 个全外显子样本。Atlas2 套件可在 http://sourceforge.net/projects/atlas2/ 下载。除了命令行版本外,该套件还已集成到 Genboree Workbench 中,允许具有最少信息学专业知识的生物医学科学家通过简单的 Web 界面远程调用、查看和进一步分析变异。通过 Genboree 浏览器显示的现有基因组数据库也简化了从变异发现到功能基因组分析的过程,为更广泛的社区提供了即用型工具包。