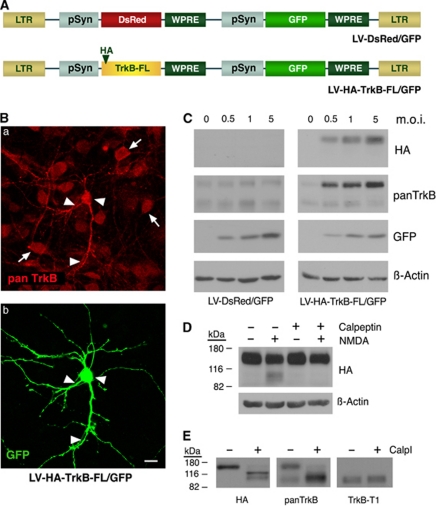
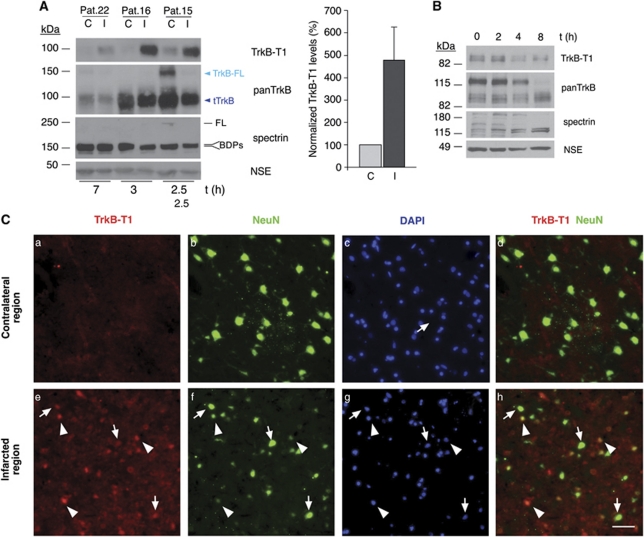
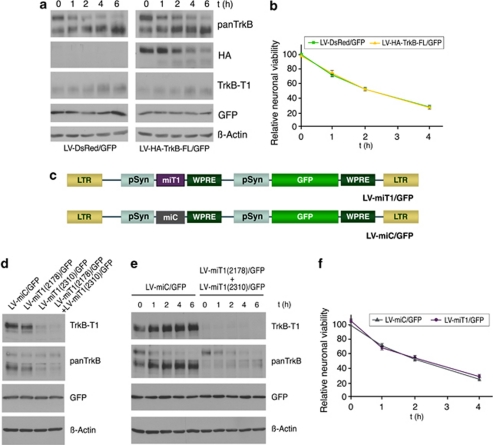
Instituto de Investigaciones Biomédicas Alberto Sols, CSIC-UAM, Madrid, Spain.
Cell Death Dis. 2012 Jan 19;3(1):e256. doi: 10.1038/cddis.2011.143.
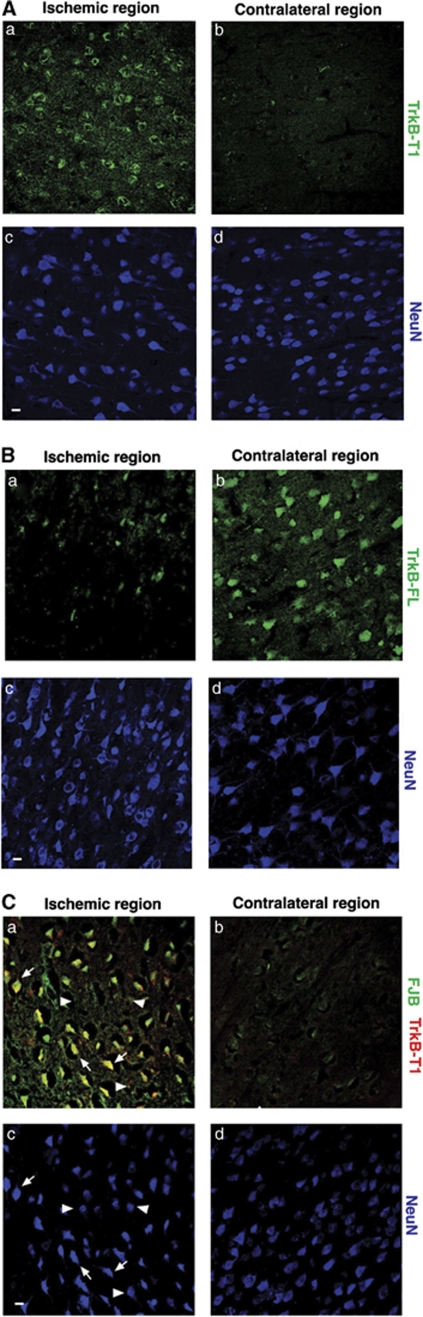
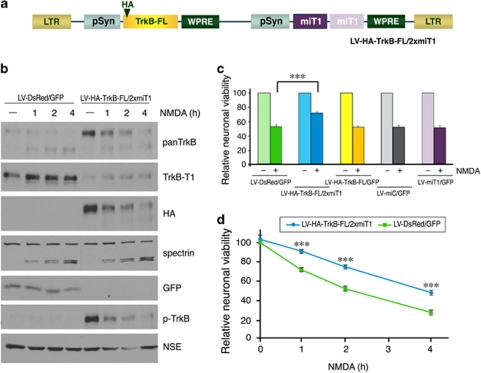
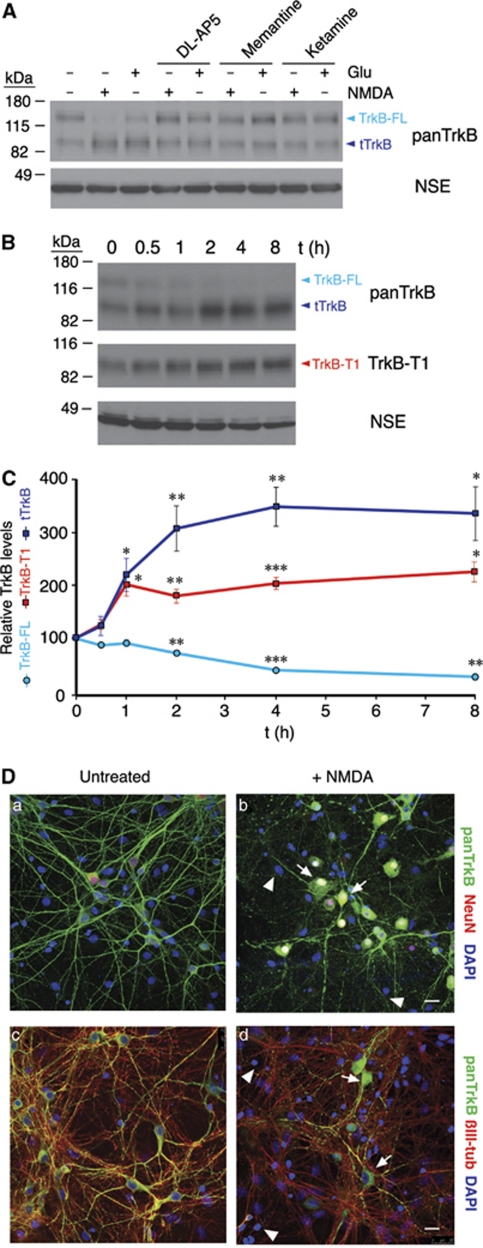
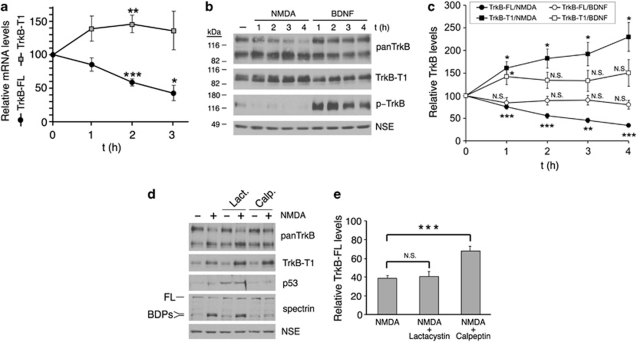
A better understanding of the mechanisms underlying neuronal death in cerebral ischemia is required for the development of stroke therapies. Here we analyze the contribution of the tropomyosin-related kinase B (TrkB) neurotrophin receptor to excitotoxicity, a primary pathological mechanism in ischemia, which is induced by overstimulation of glutamate receptors of the N-methyl-D-aspartate type. We demonstrate a significant modification of TrkB expression that is strongly associated with neurodegeneration in models of ischemia and in vitro excitotoxicity. Two mechanisms cooperate for TrkB dysregulation: (1) calpain-processing of full-length TrkB (TrkB-FL), high-affinity receptor for brain-derived neurotrophic factor, which produces a truncated protein lacking the tyrosine-kinase domain and strikingly similar to the inactive TrkB-T1 isoform and (2) reverse regulation of the mRNA of these isoforms. Collectively, excitotoxicity results in a decrease of TrkB-FL, the production of truncated TrkB-FL and the upregulation of TrkB-T1. A similar neuro-specific increase of the TrkB-T1 isoform is also observed in stroke patients. A lentivirus designed for both neuro-specific TrkB-T1 interference and increased TrkB-FL expression allows recovery of the TrkB-FL/TrkB-T1 balance and protects neurons from excitotoxic death. These data implicate a combination of TrkB-FL downregulation and TrkB-T1 upregulation as significant causes of neuronal death in excitotoxicity, and reveal novel targets for the design of stroke therapies.
为了开发中风治疗方法,我们需要更好地了解脑缺血中神经元死亡的机制。在这里,我们分析原肌球蛋白相关激酶 B (TrkB) 神经营养因子受体在兴奋性毒性中的作用,兴奋性毒性是缺血的主要病理机制之一,由 N-甲基-D-天冬氨酸型谷氨酸受体过度刺激引起。我们证明了 TrkB 表达的显著改变与缺血和体外兴奋性毒性模型中的神经退行性变密切相关。两种机制共同作用导致 TrkB 失调:(1)钙蛋白酶处理全长 TrkB(TrkB-FL),它是脑源性神经营养因子的高亲和力受体,产生缺乏酪氨酸激酶结构域的截短蛋白,与非活性 TrkB-T1 同工型非常相似;(2)这些同工型的 mRNA 反向调节。总之,兴奋性毒性导致 TrkB-FL 减少、截短的 TrkB-FL 产生和 TrkB-T1 上调。在中风患者中也观察到类似的神经特异性 TrkB-T1 同工型增加。一种设计用于神经特异性 TrkB-T1 干扰和增加 TrkB-FL 表达的慢病毒允许恢复 TrkB-FL/TrkB-T1 平衡并保护神经元免受兴奋性毒性死亡。这些数据表明 TrkB-FL 下调和 TrkB-T1 上调的组合是兴奋性毒性中神经元死亡的重要原因,并为中风治疗方法的设计揭示了新的靶点。