Laboratory for Molecular Modeling, Division of Medicinal Chemistry and Natural Products and Carolina Exploratory Center for Cheminformatics Research, School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7360, USA.
Proteins. 2012 Jun;80(6):1503-21. doi: 10.1002/prot.24035. Epub 2012 Mar 13.
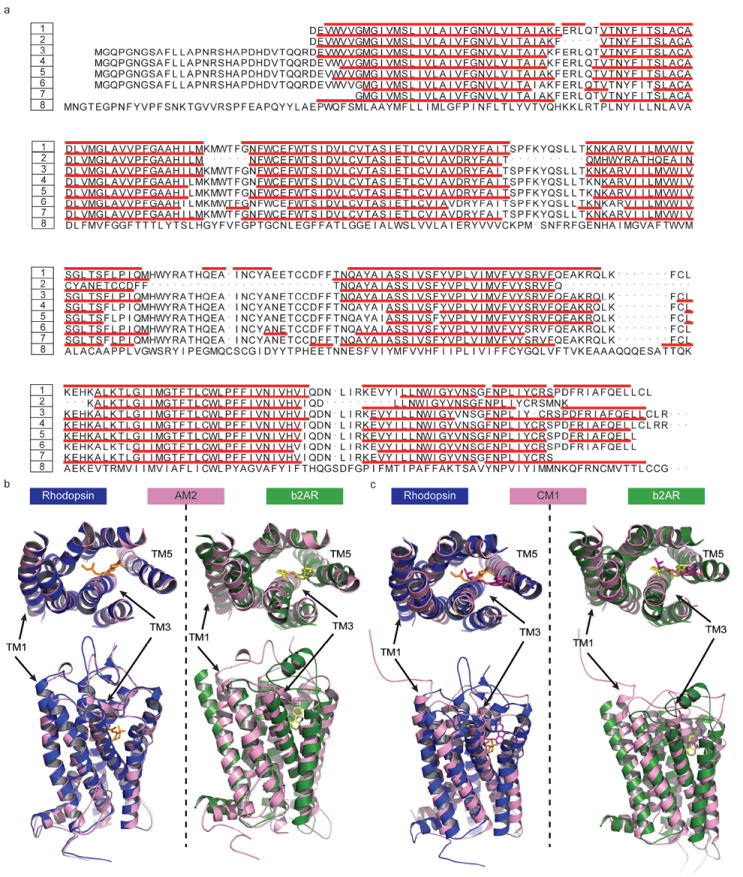
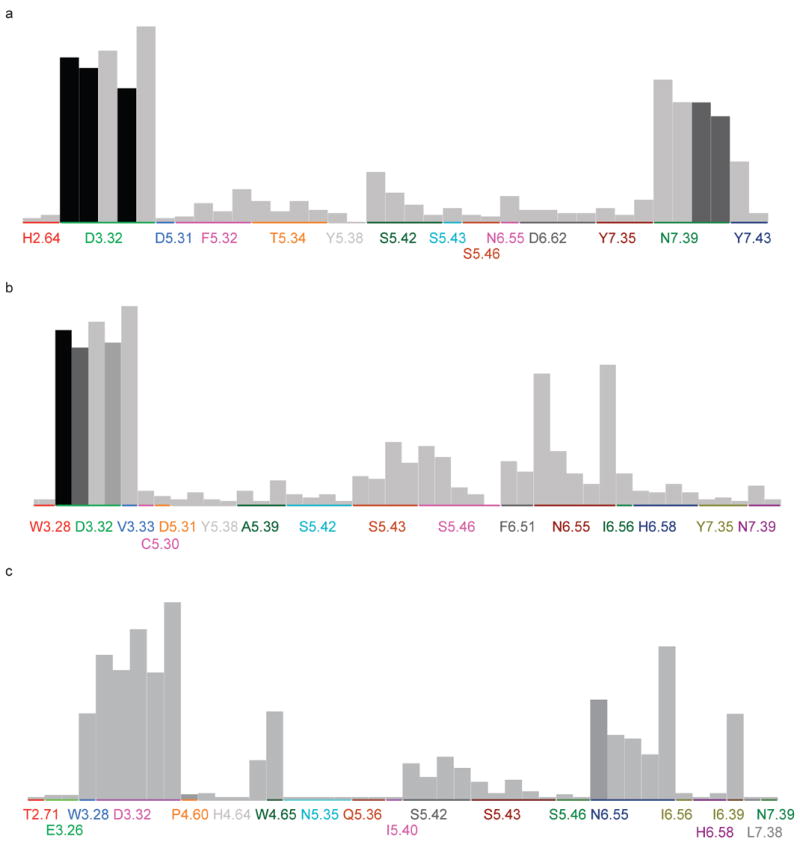
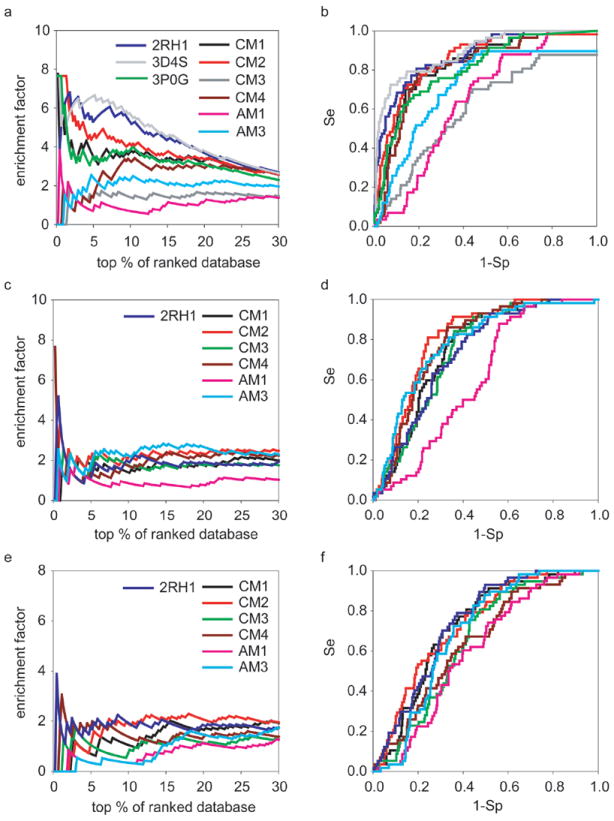
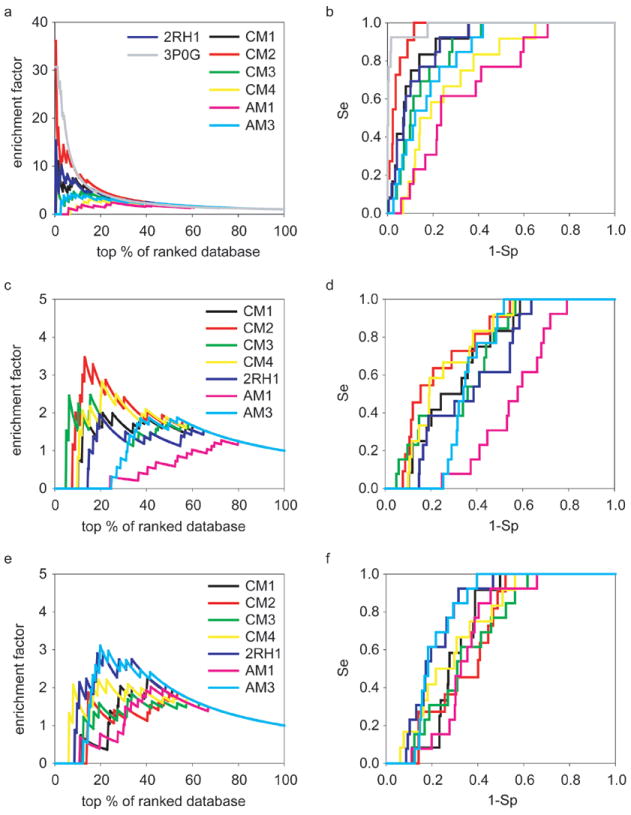
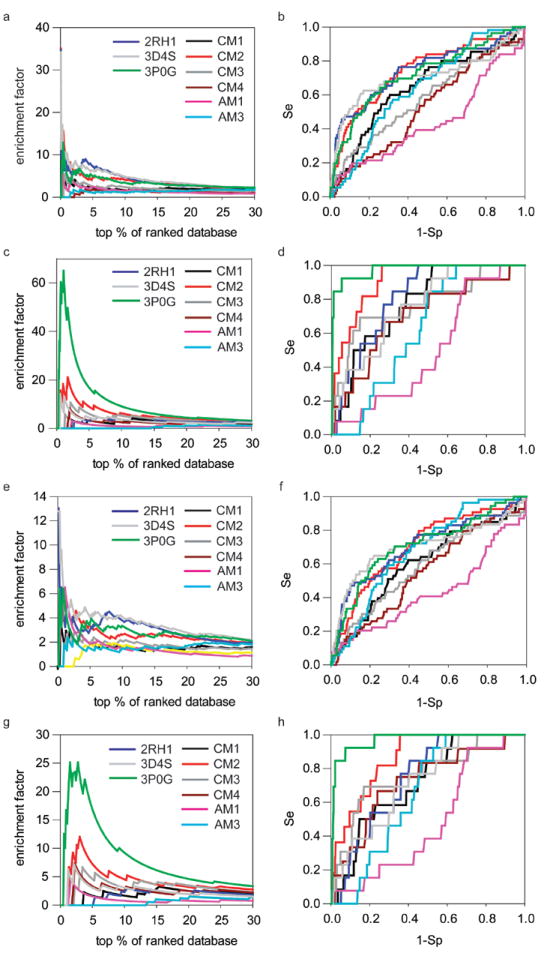
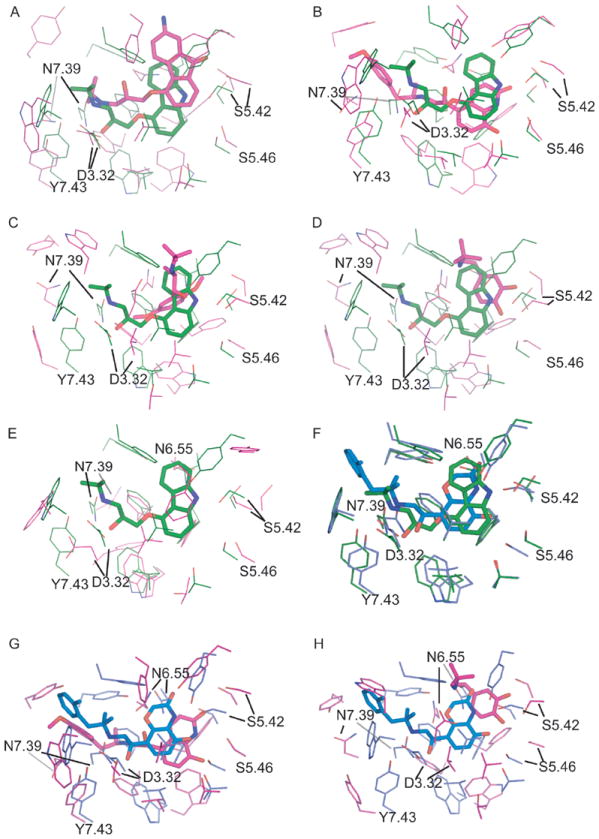
Recent highly expected structural characterizations of agonist-bound and antagonist-bound beta-2 adrenoreceptor (β2AR) by X-ray crystallography have been widely regarded as critical advances to enable more effective structure-based discovery of GPCRs ligands. It appears that this very important development may have undermined many previous efforts to develop 3D theoretical models of GPCRs. To address this question directly, we have compared several historical β2AR models versus the inactive state and nanobody-stabilized active state of β2AR crystal structures in terms of their structural similarity and effectiveness of use in virtual screening for β2AR specific agonists and antagonists. Theoretical models, incluing both homology and de novo types, were collected from five different groups who have published extensively in the field of GPCRs modeling. All models were built before X-ray structures became available. In general, β2AR theoretical models differ significantly from the crystal structure in terms of TMH definition and the global packing. Nevertheless, surprisingly, several models afforded hit rates resulting from virtual screening of large chemical library enriched by known β2AR ligands that exceeded those using X-ray structures. The hit rates were particularly higher for agonists. Furthemore, the screening performance of models is associated with local structural quality, such as the RMSDs for binding pocket residues and the ability to capture accurately, most if not all critical protein/ligand interactions. These results suggest that carefully built models of GPCRs could capture critical chemical and structural features of the binding pocket, and thus may be even more useful for practical structure-based drug discovery than X-ray structures.
最近,通过 X 射线晶体学对激动剂结合和拮抗剂结合的β-2 肾上腺素能受体(β2AR)进行的备受期待的结构特征描述被广泛认为是实现更有效的基于结构的 GPCR 配体发现的关键进展。看来,这一非常重要的发展可能破坏了许多以前开发 GPCR 三维理论模型的努力。为了直接解决这个问题,我们比较了几个历史上的β2AR 模型与β2AR 晶体结构的非活性状态和纳米体稳定的活性状态,比较它们在虚拟筛选β2AR 特异性激动剂和拮抗剂方面的结构相似性和有效性。理论模型,包括同源和从头类型,都是从五个在 GPCR 建模领域广泛发表文章的不同小组中收集的。所有模型都是在 X 射线结构可用之前构建的。一般来说,β2AR 理论模型在 TMH 定义和全局包装方面与晶体结构有很大的不同。尽管如此,令人惊讶的是,一些模型在虚拟筛选富含已知β2AR 配体的大型化学文库时,获得的命中率超过了使用 X 射线结构的命中率。对于激动剂,命中率更高。此外,模型的筛选性能与局部结构质量相关,例如结合口袋残基的 RMSD 和准确捕捉大部分(如果不是全部)关键蛋白/配体相互作用的能力。这些结果表明,精心构建的 GPCR 模型可以捕获结合口袋的关键化学和结构特征,因此对于实际的基于结构的药物发现可能比 X 射线结构更有用。