Division of Gastroenterology, Hepatology, and Nutrition, The Children's Hospital of Philadelphia, Philadelphia, PA, USA.
Mod Pathol. 2012 May;25(5):751-7. doi: 10.1038/modpathol.2011.212. Epub 2012 Feb 3.
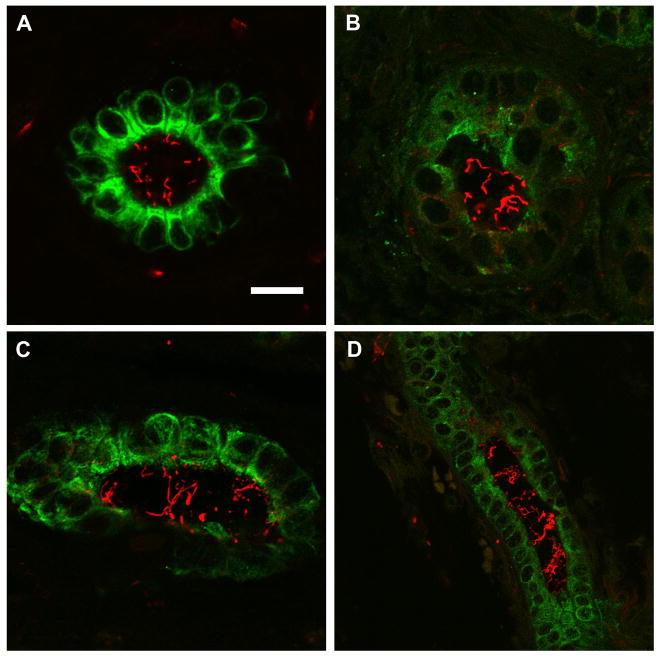
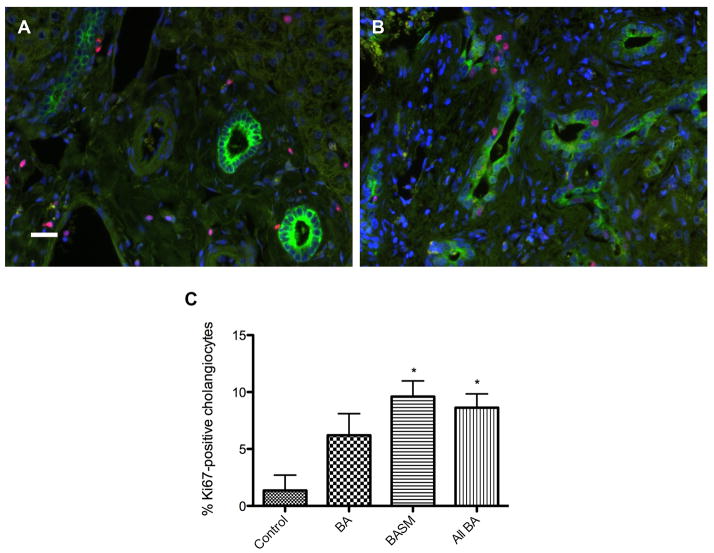
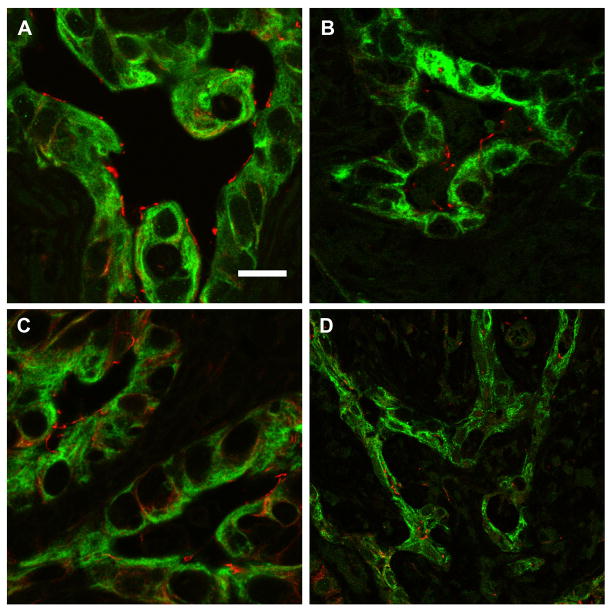
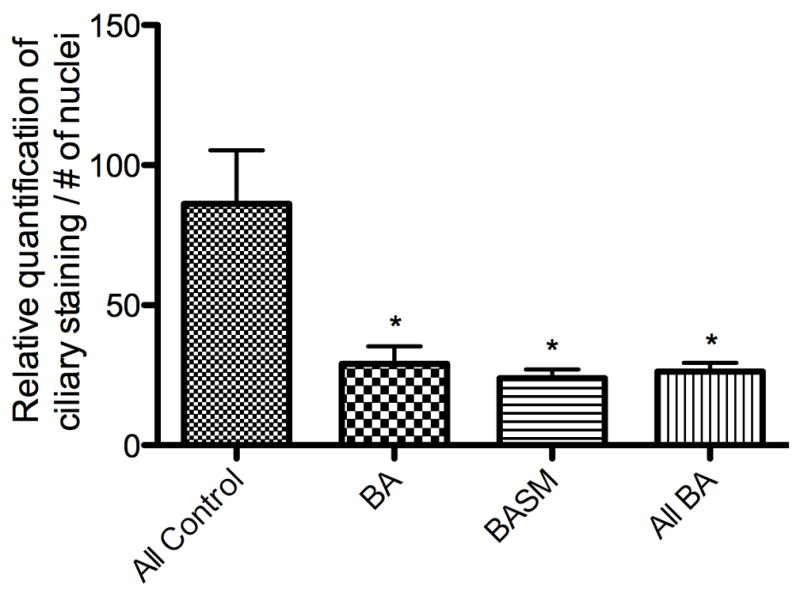
Biliary atresia (BA) is a neonatal disorder characterized by aggressive fibroinflammatory obliteration of the biliary tract. Approximately 20 percent of BA patients demonstrate left-right laterality defects (syndromic BA). Cilia participate in important physiological functions in cholangiocytes, and as some ciliopathies have been associated with both laterality defects and hepatic fibrosis, we hypothesized that patients with syndromic BA exhibit abnormalities of cholangiocyte cilia that disrupt cholangiocyte homeostasis. Nine BA specimens were studied, including pre-Kasai diagnostic biopsies (n=7) and liver explants (n=2). Five specimens were from patients with laterality defects. These were compared with normal pediatric livers, as well as livers affected by primary sclerosing cholangitis, Wilson's disease, and cardiac cirrhosis. Biopsy sections were stained with antibodies against keratin 19 (a cholangiocyte marker) and acetylated α-tubulin (a cilia marker) and were visualized by confocal microscopy. Computer-assisted relative quantification was used to compare staining of cilia within bile ducts among samples. Surprisingly, cilia in BA specimens were significantly shorter, abnormal in their orientation, and less abundant compared with normal liver and disease controls regardless of the presence of a laterality defect. There are significant abnormalities of cholangiocyte cilia in both syndromic and non-syndromic BA livers compared with normal livers and livers affected by other cholestatic diseases. Although this may result from severe cholestasis or inflammation, it may also reflect common mechanistic pathways in different forms of BA and may have important implications for understanding the progression of the disease.
先天性胆道闭锁(BA)是一种新生儿疾病,其特征为胆道进行性纤维炎症性闭塞。约 20%的 BA 患者存在左右侧位缺陷(综合征性 BA)。纤毛参与胆管细胞的重要生理功能,一些纤毛病与侧位缺陷和肝纤维化有关,因此我们假设综合征性 BA 患者的胆管细胞纤毛存在异常,破坏胆管细胞的稳态。研究了 9 个 BA 标本,包括术前 Kasai 诊断活检(n=7)和肝移植标本(n=2)。5 个标本来自侧位缺陷患者。将这些标本与正常儿童肝脏以及原发性硬化性胆管炎、Wilson 病和心脏肝硬化肝脏进行比较。用针对角蛋白 19(胆管细胞标志物)和乙酰化α-微管蛋白(纤毛标志物)的抗体对活检切片进行染色,并通过共聚焦显微镜进行观察。采用计算机辅助相对定量方法比较样本中胆管内纤毛的染色。令人惊讶的是,与正常肝脏和疾病对照相比,BA 标本中的纤毛明显更短、取向异常且数量减少,无论是否存在侧位缺陷。与正常肝脏和其他胆汁淤积性疾病的肝脏相比,综合征性和非综合征性 BA 肝脏中的胆管细胞纤毛均存在明显异常。尽管这可能是由于严重的胆汁淤积或炎症所致,但也可能反映了不同形式的 BA 中存在共同的机制途径,这对理解疾病的进展具有重要意义。