Department of Microbiology and Molecular Genetics, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania 15213, USA.
J Biol Chem. 2012 Apr 6;287(15):12036-49. doi: 10.1074/jbc.M111.307058. Epub 2012 Feb 10.
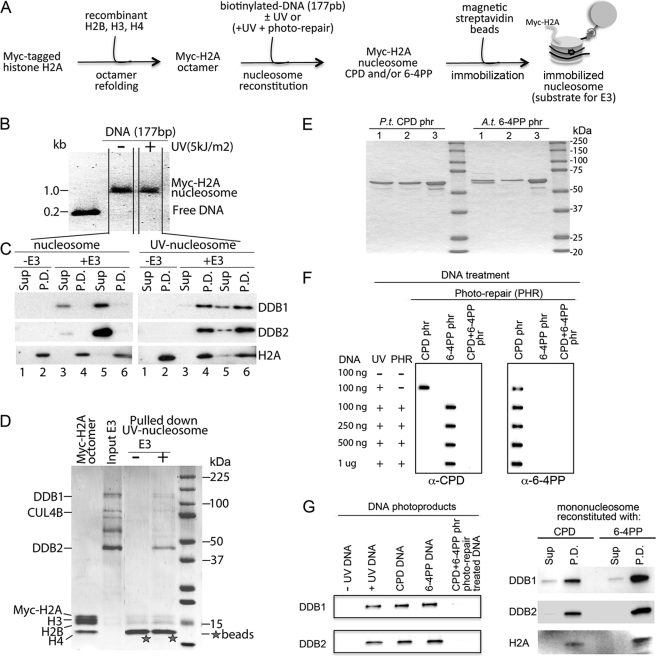
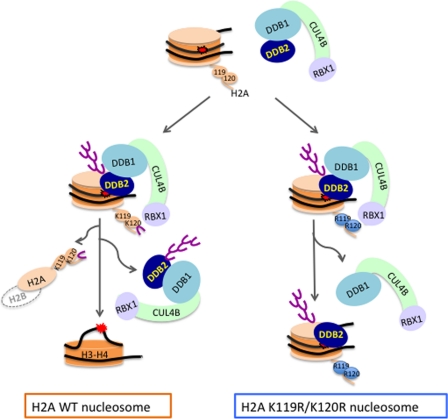
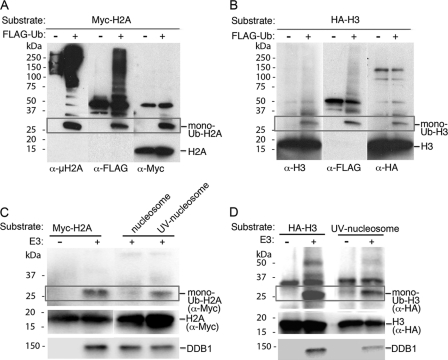
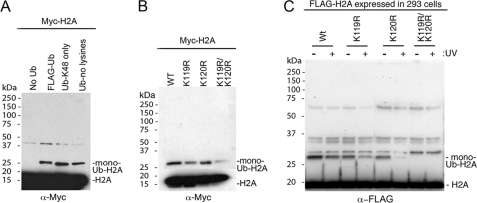
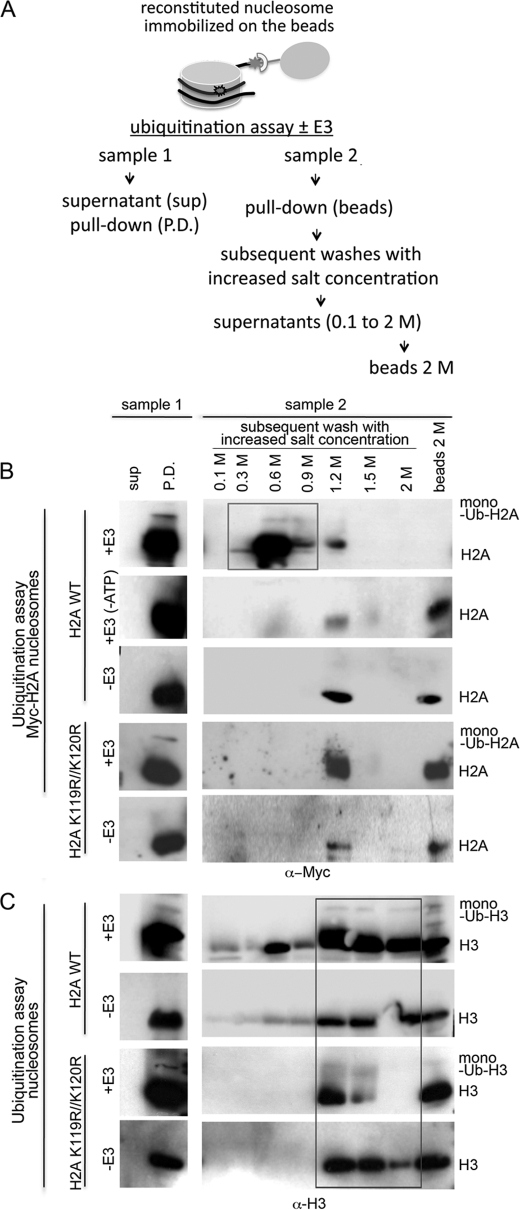
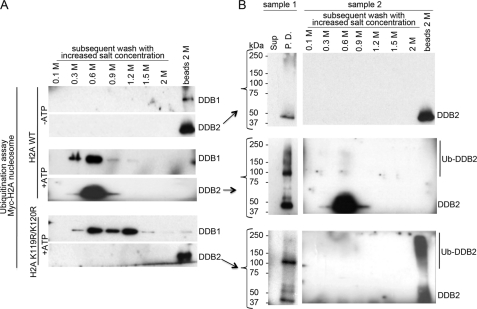
How the nucleotide excision repair (NER) machinery gains access to damaged chromatinized DNA templates and how the chromatin structure is modified to promote efficient repair of the non-transcribed genome remain poorly understood. The UV-damaged DNA-binding protein complex (UV-DDB, consisting of DDB1 and DDB2, the latter of which is mutated in xeroderma pigmentosum group E patients, is a substrate-recruiting module of the cullin 4B-based E3 ligase complex, DDB1-CUL4B(DDB2). We previously reported that the deficiency of UV-DDB E3 ligases in ubiquitinating histone H2A at UV-damaged DNA sites in the xeroderma pigmentosum group E cells contributes to the faulty NER in these skin cancer-prone patients. Here, we reveal the mechanism by which monoubiquitination of specific H2A lysine residues alters nucleosomal dynamics and subsequently initiates NER. We show that DDB1-CUL4B(DDB2) E3 ligase specifically binds to mononucleosomes assembled with human recombinant histone octamers and nucleosome-positioning DNA containing cyclobutane pyrimidine dimers or 6-4 photoproducts photolesions. We demonstrate functionally that ubiquitination of H2A Lys-119/Lys-120 is necessary for destabilization of nucleosomes and concomitant release of DDB1-CUL4B(DDB2) from photolesion-containing DNA. Nucleosomes in which these lysines are replaced with arginines are resistant to such structural changes, and arginine mutants prevent the eviction of H2A and dissociation of polyubiquitinated DDB2 from UV-damaged nucleosomes. The partial eviction of H3 from the nucleosomes is dependent on ubiquitinated H2A Lys-119/Lys-120. Our results provide mechanistic insight into how post-translational modification of H2A at the site of a photolesion initiates the repair process and directly affects the stability of the human genome.
核苷酸切除修复(NER)机制如何获得受损的染色质化 DNA 模板,以及染色质结构如何修饰以促进非转录基因组的有效修复,这些仍然知之甚少。UV 损伤 DNA 结合蛋白复合物(UV-DDB,由 DDB1 和 DDB2 组成,后者在 Xeroderma pigmentosum 组 E 患者中发生突变)是基于 Cullin 4B 的 E3 连接酶复合物 DDB1-CUL4B(DDB2)的底物募集模块。我们之前报道,Xeroderma pigmentosum 组 E 细胞中 UV-DDB E3 连接酶缺乏对 UV 损伤 DNA 位点的组蛋白 H2A 泛素化导致这些皮肤癌易感患者的 NER 错误。在这里,我们揭示了特定 H2A 赖氨酸残基单泛素化改变核小体动力学并随后启动 NER 的机制。我们表明,DDB1-CUL4B(DDB2)E3 连接酶特异性结合与人重组组蛋白八聚体组装的单核小体和含有环丁烷嘧啶二聚体或 6-4 光产物光损伤的核小体位点 DNA。我们功能证明 H2A Lys-119/Lys-120 的泛素化对于核小体的不稳定以及 DDB1-CUL4B(DDB2)从含有光损伤的 DNA 上的释放是必需的。这些赖氨酸被精氨酸取代的核小体对这种结构变化具有抗性,并且精氨酸突变体阻止 H2A 的逐出和多泛素化 DDB2 从 UV 损伤核小体上的解离。核小体中 H3 的部分逐出依赖于泛素化的 H2A Lys-119/Lys-120。我们的结果为组蛋白 H2A 在光损伤部位的翻译后修饰如何启动修复过程并直接影响人类基因组的稳定性提供了机制上的见解。