Department of Veterinary Pathobiology, Texas A&M University College of Veterinary Medicine and Biomedical Sciences, College Station, TX, USA.
BMC Genomics. 2012 Feb 17;13:78. doi: 10.1186/1471-2164-13-78.
The catalog of genetic variants in the horse genome originates from a few select animals, the majority originating from the Thoroughbred mare used for the equine genome sequencing project. The purpose of this study was to identify genetic variants, including single nucleotide polymorphisms (SNPs), insertion/deletion polymorphisms (INDELs), and copy number variants (CNVs) in the genome of an individual Quarter Horse mare sequenced by next-generation sequencing.
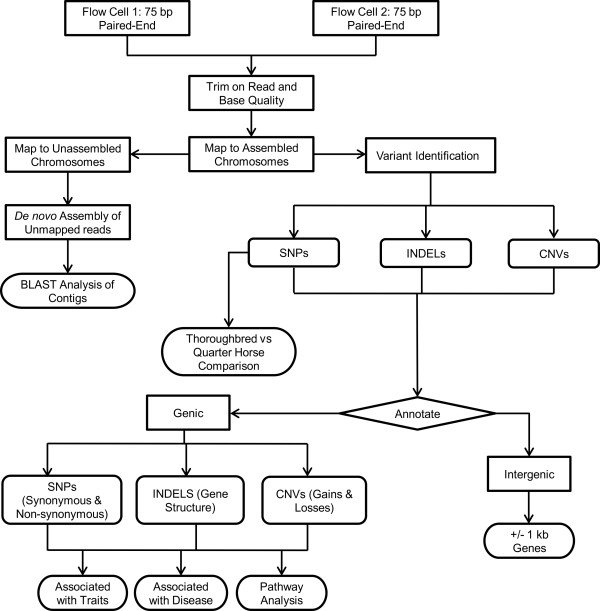
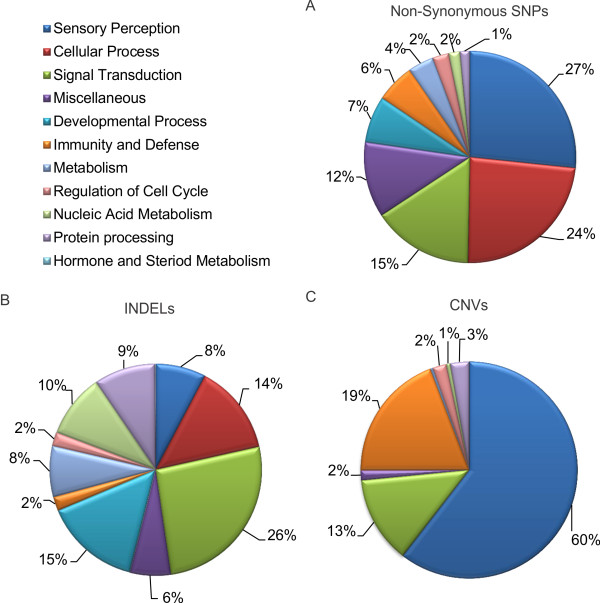
Using massively parallel paired-end sequencing, we generated 59.6 Gb of DNA sequence from a Quarter Horse mare resulting in an average of 24.7X sequence coverage. Reads were mapped to approximately 97% of the reference Thoroughbred genome. Unmapped reads were de novo assembled resulting in 19.1 Mb of new genomic sequence in the horse. Using a stringent filtering method, we identified 3.1 million SNPs, 193 thousand INDELs, and 282 CNVs. Genetic variants were annotated to determine their impact on gene structure and function. Additionally, we genotyped this Quarter Horse for mutations of known diseases and for variants associated with particular traits. Functional clustering analysis of genetic variants revealed that most of the genetic variation in the horse's genome was enriched in sensory perception, signal transduction, and immunity and defense pathways.
This is the first sequencing of a horse genome by next-generation sequencing and the first genomic sequence of an individual Quarter Horse mare. We have increased the catalog of genetic variants for use in equine genomics by the addition of novel SNPs, INDELs, and CNVs. The genetic variants described here will be a useful resource for future studies of genetic variation regulating performance traits and diseases in equids.
马基因组中的遗传变异目录来源于少数精选动物,其中大多数来源于用于马基因组测序项目的纯血母马。本研究的目的是鉴定个体夸特马基因组中的遗传变异,包括单核苷酸多态性(SNPs)、插入/缺失多态性(INDELs)和拷贝数变异(CNVs)。
使用大规模平行配对末端测序,我们从一匹夸特马中生成了 59.6 Gb 的 DNA 序列,平均序列覆盖度为 24.7X。读取序列被映射到大约 97%的参考纯血马基因组。未映射的读取进行从头组装,导致马的基因组中产生了 19.1 Mb 的新基因组序列。使用严格的过滤方法,我们鉴定出了 310 万个 SNPs、19.3 万个 INDELs 和 282 个 CNVs。遗传变异被注释以确定它们对基因结构和功能的影响。此外,我们对这匹夸特马进行了基因突变的检测,包括已知疾病的突变和与特定特征相关的变异。遗传变异的功能聚类分析表明,马基因组中的大部分遗传变异富集在感觉感知、信号转导和免疫与防御途径中。
这是首次使用下一代测序对马基因组进行测序,也是首次对个体夸特马母马进行基因组测序。我们通过增加新的 SNPs、INDELs 和 CNVs,增加了用于马基因组学的遗传变异目录。这里描述的遗传变异将成为未来研究调控马属动物性能特征和疾病的遗传变异的有用资源。