Lin Wan-Yu, Zhang Boshao, Yi Nengjun, Gao Guimin, Liu Nianjun
Department of Biostatistics, University of Alabama at Birmingham, 1665 University Boulevard, Birmingham, AL 35294, USA.
BMC Proc. 2011 Nov 29;5 Suppl 9(Suppl 9):S118. doi: 10.1186/1753-6561-5-S9-S118.
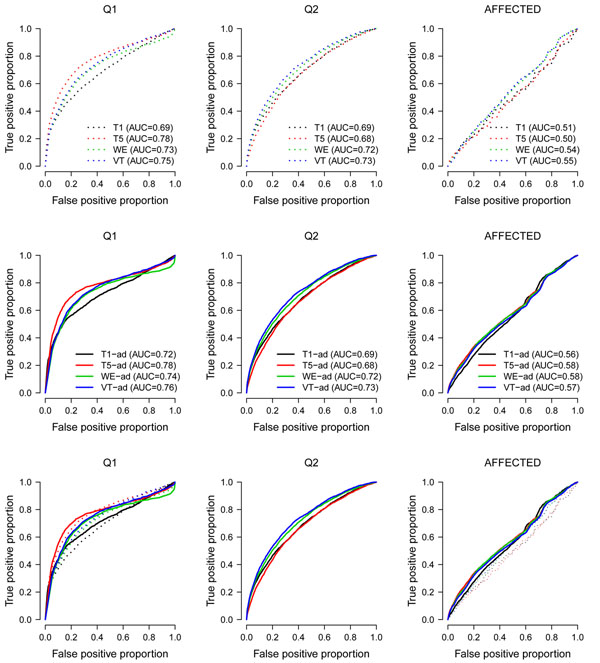
Genome-wide association studies have successfully identified many common variants associated with complex human diseases. However, a large portion of the remaining heritability cannot be explained by these common variants. Exploring rare variants associated with diseases is now catching more attention. Several methods have been recently proposed for identification of rare variants. Among them, the fixed-threshold, weighted-sum, and variable-threshold methods are effective in combining the information of multiple variants into a functional unit; these approaches are commonly used. We evaluate the performance of these three methods. Based on our analyses of the Genetic Analysis Workshop 17 data, we find that no method is universally better than the others. Furthermore, adjusting for potential covariates can not only increase the true-positive proportions but also reduce the false-positive proportions. Our study concludes that there is no uniformly most powerful test among the three methods we compared (the fixed-threshold, weighted-sum, and variable-threshold methods), and their performances depend on the underlying genetic architecture of a disease.
全基因组关联研究已成功识别出许多与复杂人类疾病相关的常见变异。然而,其余很大一部分遗传力无法用这些常见变异来解释。探索与疾病相关的罕见变异正受到更多关注。最近提出了几种识别罕见变异的方法。其中,固定阈值法、加权和法以及可变阈值法在将多个变异的信息整合为一个功能单元方面很有效;这些方法被广泛使用。我们评估了这三种方法的性能。基于我们对遗传分析研讨会17数据的分析,我们发现没有一种方法普遍优于其他方法。此外,对潜在协变量进行调整不仅可以提高真阳性比例,还可以降低假阳性比例。我们的研究得出结论,在我们比较的三种方法(固定阈值法、加权和法以及可变阈值法)中,没有一种是普遍最强大的检验方法,它们的性能取决于疾病的潜在遗传结构。