Li Gengxin, Ferguson John, Zheng Wei, Lee Joon Sang, Zhang Xianghua, Li Lun, Kang Jia, Yan Xiting, Zhao Hongyu
Department of Epidemiology and Public Health, Yale University, 60 College Street, New Haven, CT 06520, USA.
BMC Proc. 2011 Nov 29;5 Suppl 9(Suppl 9):S46. doi: 10.1186/1753-6561-5-S9-S46.
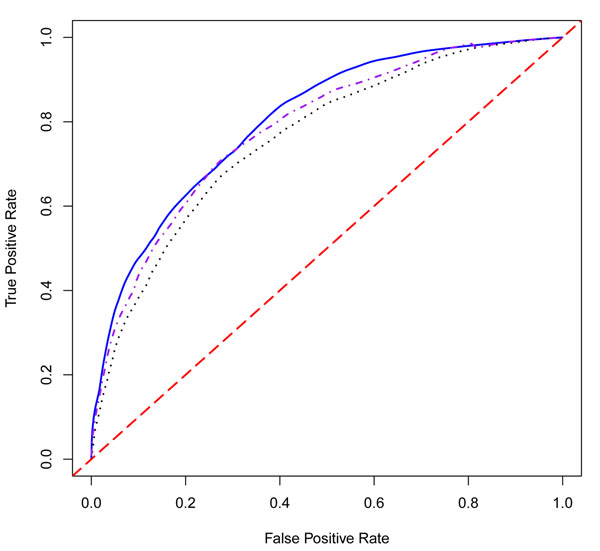
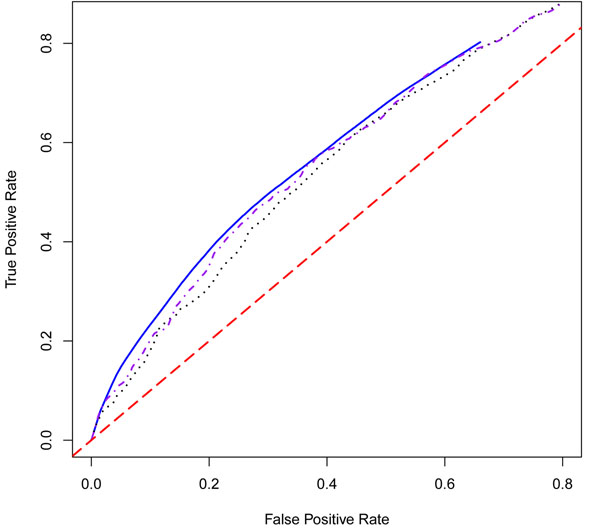
We consider the application of Efron's empirical Bayes classification method to risk prediction in a genome-wide association study using the Genetic Analysis Workshop 17 (GAW17) data. A major advantage of using this method is that the effect size distribution for the set of possible features is empirically estimated and that all subsequent parameter estimation and risk prediction is guided by this distribution. Here, we generalize Efron's method to allow for some of the peculiarities of the GAW17 data. In particular, we introduce two ways to extend Efron's model: a weighted empirical Bayes model and a joint covariance model that allows the model to properly incorporate the annotation information of single-nucleotide polymorphisms (SNPs). In the course of our analysis, we examine several aspects of the possible simulation model, including the identity of the most important genes, the differing effects of synonymous and nonsynonymous SNPs, and the relative roles of covariates and genes in conferring disease risk. Finally, we compare the three methods to each other and to other classifiers (random forest and neural network).
我们考虑将埃弗龙的经验贝叶斯分类方法应用于利用遗传分析研讨会17(GAW17)数据进行的全基因组关联研究中的风险预测。使用该方法的一个主要优点是,对一组可能特征的效应大小分布进行了经验估计,并且所有后续的参数估计和风险预测都由该分布指导。在这里,我们对埃弗龙的方法进行了推广,以适应GAW17数据的一些特点。特别是,我们引入了两种扩展埃弗龙模型的方法:加权经验贝叶斯模型和联合协方差模型,该模型允许模型正确纳入单核苷酸多态性(SNP)的注释信息。在我们的分析过程中,我们研究了可能的模拟模型的几个方面,包括最重要基因的身份、同义SNP和非同义SNP的不同效应,以及协变量和基因在赋予疾病风险方面的相对作用。最后,我们将这三种方法相互比较,并与其他分类器(随机森林和神经网络)进行比较。