Kumar Chetan, Choudhary Alok
Department of Electrical Engineering and Computer Science, Northwestern University, Evanston, IL 60201, USA.
EURASIP J Bioinform Syst Biol. 2012 Feb 29;2012(1):1. doi: 10.1186/1687-4153-2012-1.
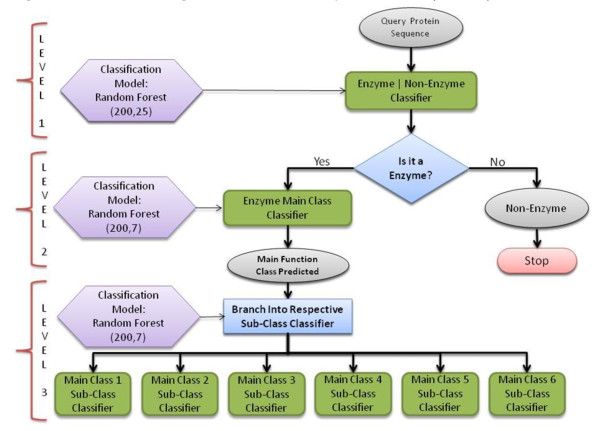
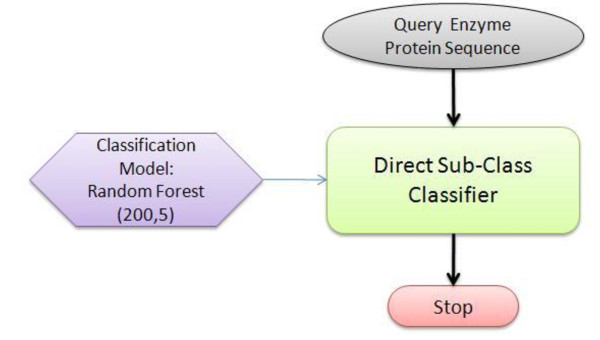
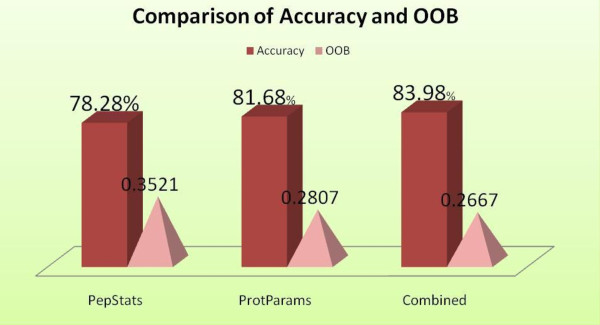
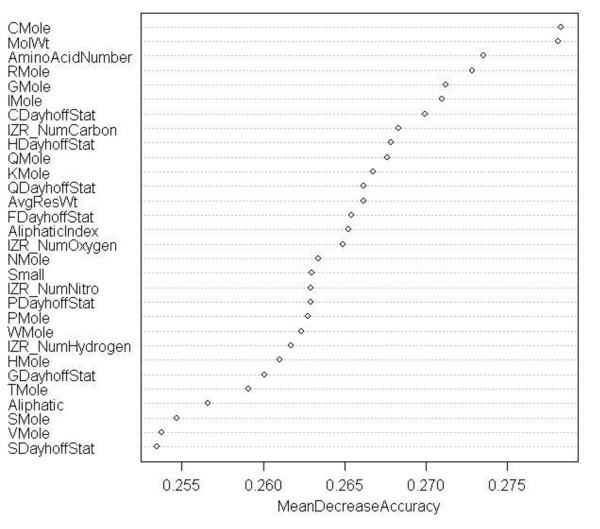
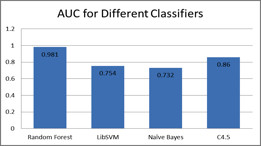
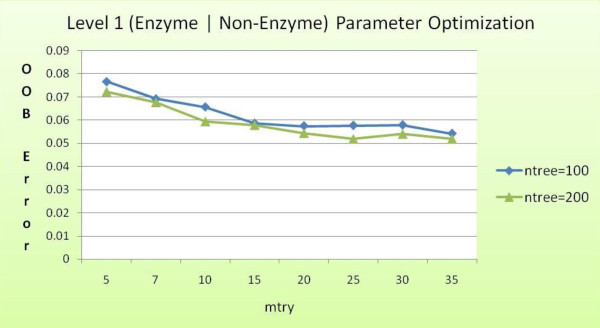
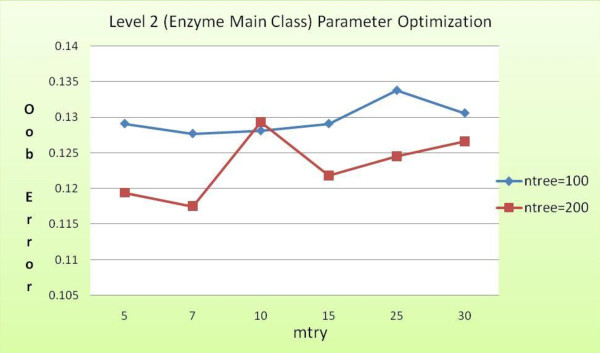
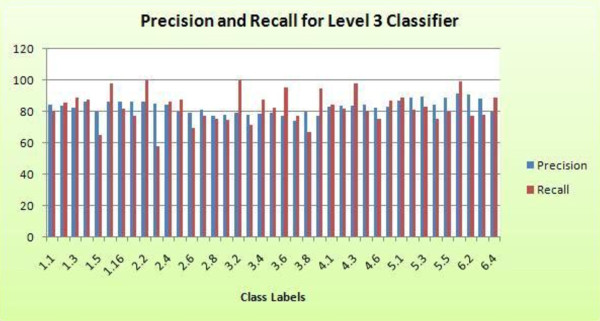
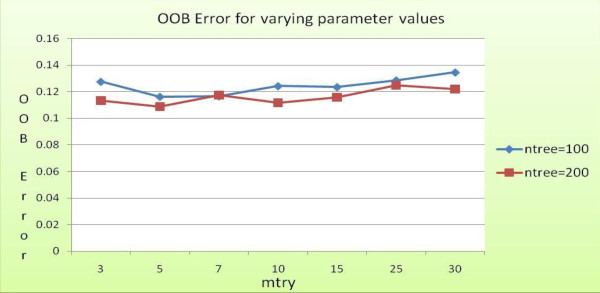
Advancements in sequencing technologies have witnessed an exponential rise in the number of newly found enzymes. Enzymes are proteins that catalyze bio-chemical reactions and play an important role in metabolic pathways. Commonly, function of such enzymes is determined by experiments that can be time consuming and costly. Hence, a need for a computing method is felt that can distinguish protein enzyme sequences from those of non-enzymes and reliably predict the function of the former. To address this problem, approaches that cluster enzymes based on their sequence and structural similarity have been presented. But, these approaches are known to fail for proteins that perform the same function and are dissimilar in their sequence and structure. In this article, we present a supervised machine learning model to predict the function class and sub-class of enzymes based on a set of 73 sequence-derived features. The functional classes are as defined by International Union of Biochemistry and Molecular Biology. Using an efficient data mining algorithm called random forest, we construct a top-down three layer model where the top layer classifies a query protein sequence as an enzyme or non-enzyme, the second layer predicts the main function class and bottom layer further predicts the sub-function class. The model reported overall classification accuracy of 94.87% for the first level, 87.7% for the second, and 84.25% for the bottom level. Our results compare very well with existing methods, and in many cases report better performance. Using feature selection methods, we have shown the biological relevance of a few of the top rank attributes.
测序技术的进步见证了新发现酶的数量呈指数级增长。酶是催化生物化学反应并在代谢途径中发挥重要作用的蛋白质。通常,此类酶的功能是通过可能耗时且成本高昂的实验来确定的。因此,人们感到需要一种计算方法,该方法可以区分蛋白质酶序列和非酶序列,并可靠地预测前者的功能。为了解决这个问题,已经提出了基于酶的序列和结构相似性对其进行聚类的方法。但是,已知这些方法对于执行相同功能但序列和结构不同的蛋白质会失效。在本文中,我们提出了一种监督机器学习模型,用于基于一组73个源自序列的特征来预测酶的功能类别和子类别。功能类别由国际生物化学与分子生物学联盟定义。使用一种称为随机森林的高效数据挖掘算法,我们构建了一个自上而下的三层模型,其中顶层将查询蛋白质序列分类为酶或非酶,第二层预测主要功能类别,底层进一步预测子功能类别。该模型报告的第一级总体分类准确率为94.87%,第二级为87.7%,底层为84.25%。我们的结果与现有方法相比非常出色,并且在许多情况下表现更好。使用特征选择方法,我们已经展示了一些顶级属性的生物学相关性。