Department of Neurological, Neuropsychological, Morphological and Motor Sciences, University of Verona, Verona, Italy.
PLoS One. 2012;7(2):e32382. doi: 10.1371/journal.pone.0032382. Epub 2012 Feb 23.
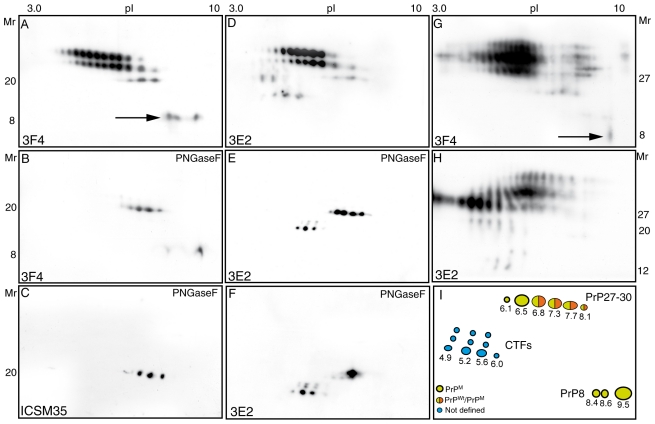
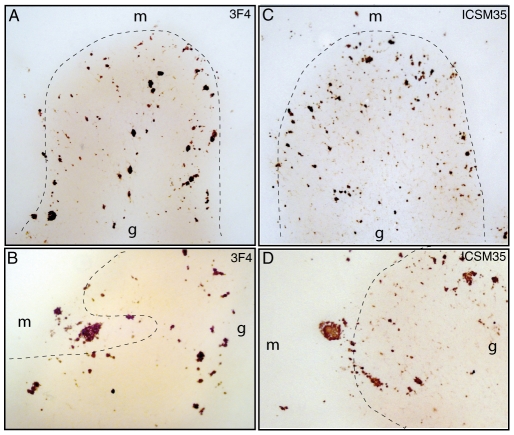
Gerstmann-Sträussler-Scheinker (GSS) disease is a dominantly inherited prion disease associated with point mutations in the Prion Protein gene. The most frequent mutation associated with GSS involves a proline-to-leucine substitution at residue 102 of the prion protein, and is characterized by marked variability at clinical, pathological and molecular levels. Previous investigations of GSS P102L have shown that disease-associated pathological prion protein, or PrP(Sc), consists of two main conformers, which under exogenous proteolysis generates a core fragment of 21 kDa and an internal fragment of 8 kDa. Both conformers are detected in subjects with spongiform degeneration, whereas only the 8 kDa fragment is recovered in cases lacking spongiosis. Several studies have reported an exclusive derivation of protease-resistant PrP(Sc) isoforms from the mutated allele; however, more recently, the propagation of protease-resistant wild-type PrP(Sc) has been described. Here we analyze the molecular and pathological phenotype of six GSS P102L cases characterized by the presence of 21 and 8 kDa PrP fragments and two subjects with only the 8 kDa PrP fragment. Using sensitive protein separation techniques and Western blots with antibodies differentially recognizing wild-type and mutant PrP we observed a range of PrP(Sc) allelic conformers, either resistant or sensitive to protease treatment in all investigated subjects. Additionally, tissue deposition of protease-sensitive wild-type PrP(Sc) molecules was seen by conventional PrP immunohistochemistry and paraffin-embedded tissue blot. Our findings enlarge the spectrum of conformational allelic PrP(Sc) quasispecies propagating in GSS P102L thus providing a molecular support to the spectrum of disease phenotypes, and, in addition, impact the diagnostic role of PrP immunohistochemistry in prion diseases.
格斯特曼-施特劳斯勒-谢因克(Gerstmann-Sträussler-Scheinker,GSS)病是一种常染色体显性遗传朊病毒病,与朊病毒蛋白基因中的点突变有关。与 GSS 相关的最常见突变涉及朊病毒蛋白第 102 位脯氨酸到亮氨酸的取代,其在临床、病理和分子水平上具有明显的变异性。以前对 GSS P102L 的研究表明,与疾病相关的病理性朊病毒蛋白或 PrP(Sc)由两种主要构象组成,在外源蛋白酶水解下生成 21 kDa 的核心片段和 8 kDa 的内部片段。两种构象都在海绵状变性的患者中检测到,而在缺乏海绵样变性的病例中只回收 8 kDa 片段。几项研究报告了抗蛋白酶的 PrP(Sc)同工型仅源自突变等位基因;然而,最近,已描述了抗蛋白酶的野生型 PrP(Sc)的传播。在这里,我们分析了六个 GSS P102L 病例的分子和病理表型,这些病例的特征是存在 21 kDa 和 8 kDa PrP 片段,以及两个仅存在 8 kDa PrP 片段的病例。使用敏感的蛋白质分离技术和 Western blot 分析,用抗体特异性识别野生型和突变型 PrP,我们观察到所有研究对象的 PrP(Sc)等位基因构象的范围,无论是对蛋白酶处理有抗性还是敏感。此外,通过常规 PrP 免疫组织化学和石蜡包埋组织印迹,观察到组织中沉积的蛋白酶敏感的野生型 PrP(Sc)分子。我们的发现扩大了 GSS P102L 中传播的构象等位基因 PrP(Sc)准种的范围,从而为疾病表型的范围提供了分子支持,此外,还影响了朊病毒病中 PrP 免疫组织化学的诊断作用。