G3 (Bethesda). 2011 Nov;1(6):457-70. doi: 10.1534/g3.111.001198. Epub 2011 Nov 1.
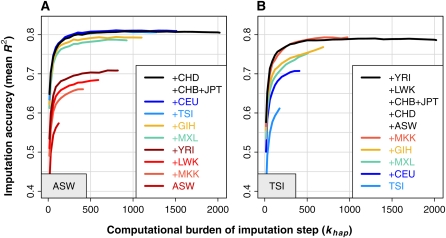
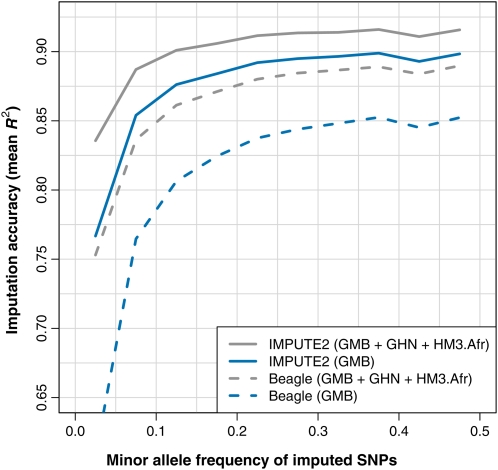
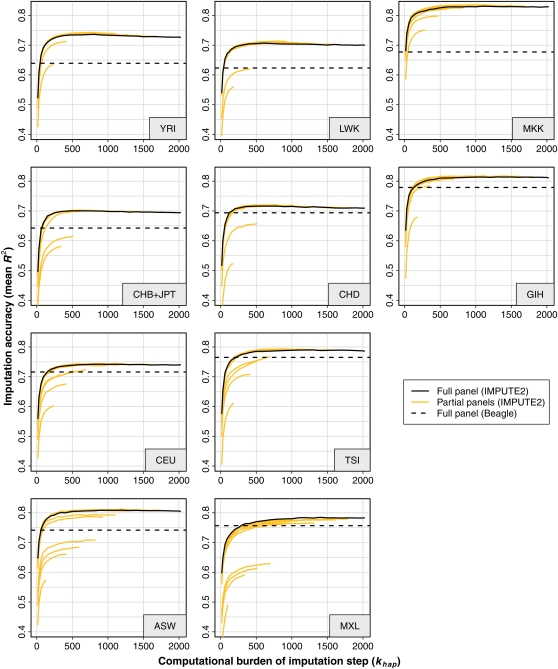
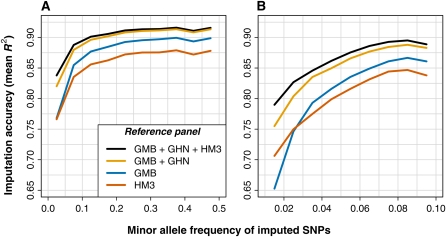
Genotype imputation is a statistical technique that is often used to increase the power and resolution of genetic association studies. Imputation methods work by using haplotype patterns in a reference panel to predict unobserved genotypes in a study dataset, and a number of approaches have been proposed for choosing subsets of reference haplotypes that will maximize accuracy in a given study population. These panel selection strategies become harder to apply and interpret as sequencing efforts like the 1000 Genomes Project produce larger and more diverse reference sets, which led us to develop an alternative framework. Our approach is built around a new approximation that uses local sequence similarity to choose a custom reference panel for each study haplotype in each region of the genome. This approximation makes it computationally efficient to use all available reference haplotypes, which allows us to bypass the panel selection step and to improve accuracy at low-frequency variants by capturing unexpected allele sharing among populations. Using data from HapMap 3, we show that our framework produces accurate results in a wide range of human populations. We also use data from the Malaria Genetic Epidemiology Network (MalariaGEN) to provide recommendations for imputation-based studies in Africa. We demonstrate that our approximation improves efficiency in large, sequence-based reference panels, and we discuss general computational strategies for modern reference datasets. Genome-wide association studies will soon be able to harness the power of thousands of reference genomes, and our work provides a practical way for investigators to use this rich information. New methodology from this study is implemented in the IMPUTE2 software package.
基因分型是一种统计技术,常用于提高遗传关联研究的功效和分辨率。 该方法通过在参考面板中使用单倍型模式来预测研究数据集中未观察到的基因型,并且已经提出了许多方法来选择参考单倍型的子集,以在给定的研究人群中最大化准确性。 随着测序工作(如 1000 基因组计划)产生更大和更多样化的参考集,这些面板选择策略变得更难应用和解释,这促使我们开发了一种替代框架。 我们的方法围绕着一个新的近似值构建,该近似值使用局部序列相似性为基因组的每个区域中的每个研究单倍型选择自定义参考面板。 这种近似使得使用所有可用的参考单倍型在计算上变得高效,这使我们能够绕过面板选择步骤,并通过捕获人群之间意外的等位基因共享来提高低频变体的准确性。 使用 HapMap 3 中的数据,我们表明我们的框架在广泛的人类群体中产生了准确的结果。 我们还使用来自疟疾遗传流行病学网络(MalariaGEN)的数据为非洲的基于 imputation 的研究提供建议。 我们证明我们的近似值提高了大型基于序列的参考面板的效率,并讨论了现代参考数据集的一般计算策略。 全基因组关联研究很快将能够利用数千个参考基因组的功能,我们的工作为研究人员提供了一种实用的方法来利用这种丰富的信息。 本研究中的新方法学已在 IMPUTE2 软件包中实现。