Genomic Research on Complex diseases (GRC Group), CSIR-Centre for Cellular and Molecular Biology, Hyderabad, Telangana, 500 007, India.
#5/1, 4th cross, Manjunatha Layout, Nagashettyhalli, 560094, Bengaluru, India.
Sci Rep. 2017 Jul 27;7(1):6733. doi: 10.1038/s41598-017-06905-6.
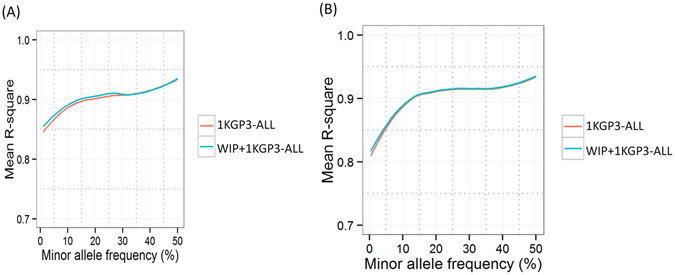
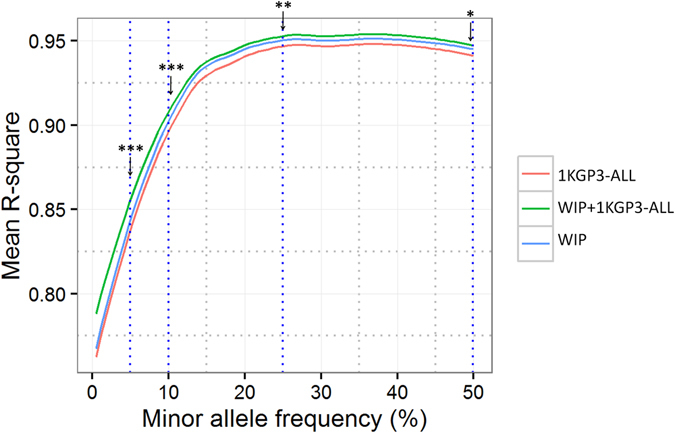
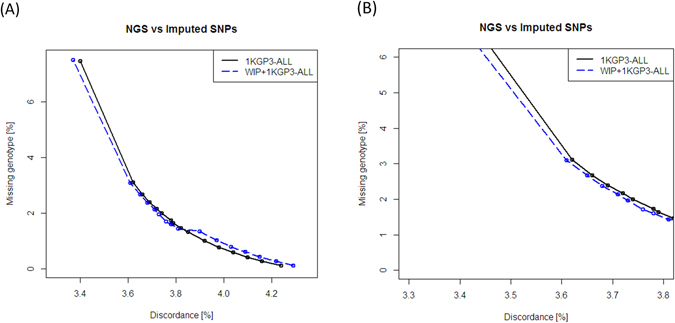
Imputation is a computational method based on the principle of haplotype sharing allowing enrichment of genome-wide association study datasets. It depends on the haplotype structure of the population and density of the genotype data. The 1000 Genomes Project led to the generation of imputation reference panels which have been used globally. However, recent studies have shown that population-specific panels provide better enrichment of genome-wide variants. We compared the imputation accuracy using 1000 Genomes phase 3 reference panel and a panel generated from genome-wide data on 407 individuals from Western India (WIP). The concordance of imputed variants was cross-checked with next-generation re-sequencing data on a subset of genomic regions. Further, using the genome-wide data from 1880 individuals, we demonstrate that WIP works better than the 1000 Genomes phase 3 panel and when merged with it, significantly improves the imputation accuracy throughout the minor allele frequency range. We also show that imputation using only South Asian component of the 1000 Genomes phase 3 panel works as good as the merged panel, making it computationally less intensive job. Thus, our study stresses that imputation accuracy using 1000 Genomes phase 3 panel can be further improved by including population-specific reference panels from South Asia.
基于单倍型共享原理的推断是一种计算方法,允许对全基因组关联研究数据集进行富集。它取决于人群的单倍型结构和基因型数据的密度。1000 基因组计划导致了推断参考面板的产生,这些面板已经在全球范围内使用。然而,最近的研究表明,特定于人群的面板可以更好地富集全基因组变体。我们比较了使用 1000 基因组计划第 3 阶段参考面板和从来自印度西部的 407 个人的全基因组数据生成的面板进行推断的准确性。使用下一代重测序数据在基因组区域的子集上交叉检查推断的变体的一致性。此外,使用来自 1880 个人的全基因组数据,我们证明 WIP 比 1000 基因组计划第 3 阶段面板更好,并且与它合并时,在整个次要等位基因频率范围内显著提高了推断准确性。我们还表明,仅使用 1000 基因组计划第 3 阶段面板的南亚成分进行推断与合并面板一样好,使其在计算上不那么密集。因此,我们的研究强调,通过包含来自南亚的特定于人群的参考面板,可以进一步提高使用 1000 基因组计划第 3 阶段面板进行推断的准确性。