The Breakthrough Breast Cancer Research Centre, The Institute of Cancer Research, London, United Kingdom.
PLoS One. 2012;7(3):e32617. doi: 10.1371/journal.pone.0032617. Epub 2012 Mar 5.
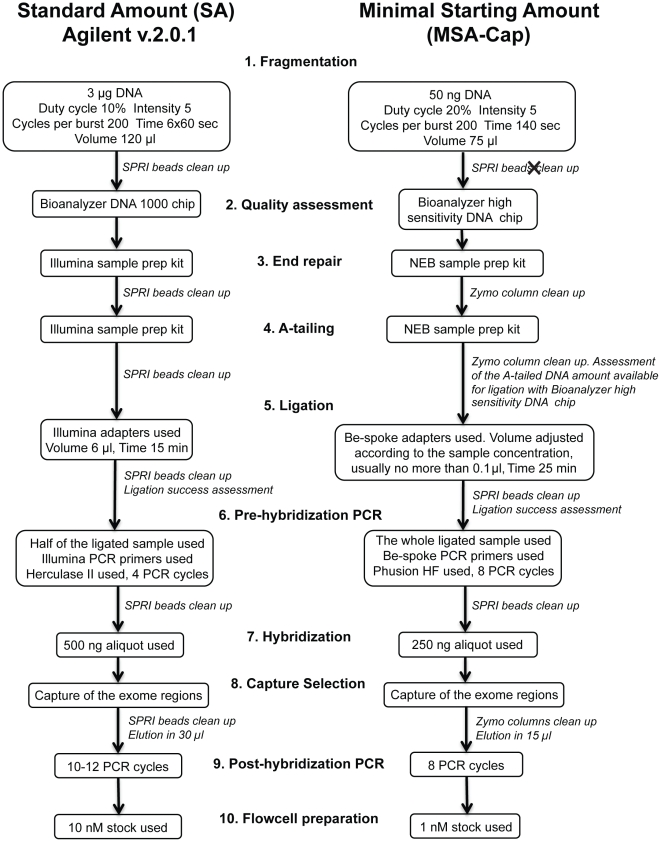
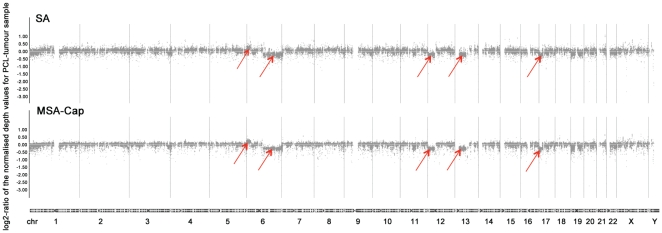
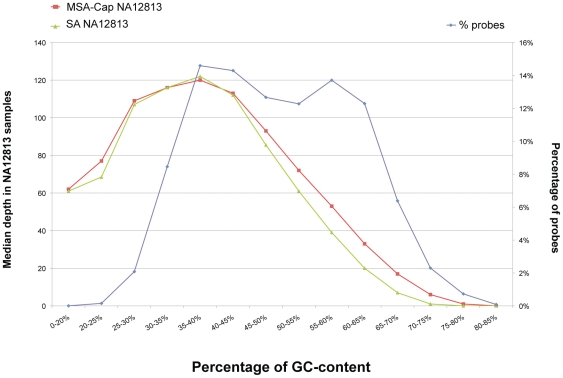
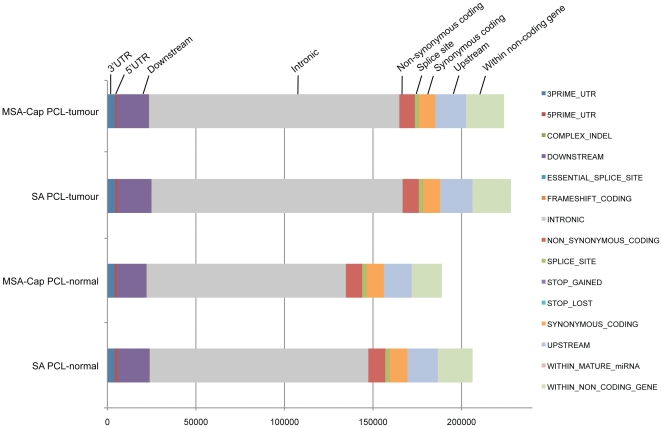
Next generation DNA sequencing (NGS) technologies have revolutionized the pace at which whole genome and exome sequences can be generated. However, despite these advances, many of the methods for targeted resequencing, such as the generation of high-depth exome sequences, are somewhat limited by the relatively large amounts of starting DNA that are normally required. In the case of tumour analysis this is particularly pertinent as many tumour biopsies often return submicrogram quantities of DNA, especially when tumours are microdissected prior to analysis. Here, we present a method for exome capture and resequencing using as little as 50 ng of starting DNA. The sequencing libraries generated by this minimal starting amount (MSA-Cap) method generate datasets that are comparable to standard amount (SA) whole exome libraries that use three micrograms of starting DNA. This method, which can be performed in most laboratories using commonly available reagents, has the potential to enhance large scale profiling efforts such as the resequencing of tumour exomes.
下一代 DNA 测序 (NGS) 技术彻底改变了全基因组和外显子组序列生成的速度。然而,尽管有了这些进步,许多靶向重测序的方法,如高通量外显子组测序的生成,仍然受到通常需要的大量起始 DNA 的限制。在肿瘤分析的情况下,这一点尤为重要,因为许多肿瘤活检通常只能返回亚微克级别的 DNA,特别是在肿瘤在分析前进行微切割时。在这里,我们提出了一种使用低至 50ng 起始 DNA 进行外显子组捕获和重测序的方法。使用这种最小起始量 (MSA-Cap) 方法生成的测序文库与使用三微克起始 DNA 的标准量 (SA) 全外显子文库生成的数据集相当。这种方法可以在大多数实验室中使用常用试剂进行,有可能增强大规模分析工作,如肿瘤外显子组的重测序。