Institute of Biophysics and Physical Biochemistry, University of Regensburg, 93040 Regensburg, Germany.
BMC Bioinformatics. 2012 Apr 5;13:55. doi: 10.1186/1471-2105-13-55.
One aim of the in silico characterization of proteins is to identify all residue-positions, which are crucial for function or structure. Several sequence-based algorithms exist, which predict functionally important sites. However, with respect to sequence information, many functionally and structurally important sites are hard to distinguish and consequently a large number of incorrectly predicted functional sites have to be expected. This is why we were interested to design a new classifier that differentiates between functionally and structurally important sites and to assess its performance on representative datasets.
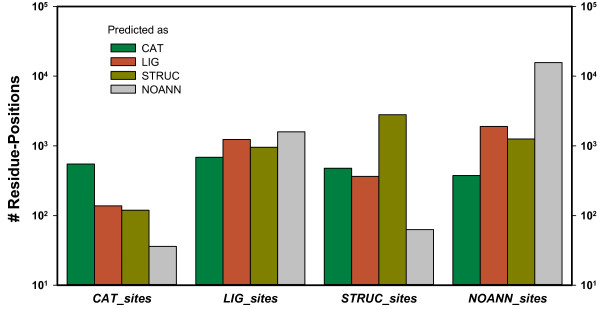
We have implemented CLIPS-1D, which predicts a role in catalysis, ligand-binding, or protein structure for residue-positions in a mutually exclusive manner. By analyzing a multiple sequence alignment, the algorithm scores conservation as well as abundance of residues at individual sites and their local neighborhood and categorizes by means of a multiclass support vector machine. A cross-validation confirmed that residue-positions involved in catalysis were identified with state-of-the-art quality; the mean MCC-value was 0.34. For structurally important sites, prediction quality was considerably higher (mean MCC = 0.67). For ligand-binding sites, prediction quality was lower (mean MCC = 0.12), because binding sites and structurally important residue-positions share conservation and abundance values, which makes their separation difficult. We show that classification success varies for residues in a class-specific manner. This is why our algorithm computes residue-specific p-values, which allow for the statistical assessment of each individual prediction. CLIPS-1D is available as a Web service at http://www-bioinf.uni-regensburg.de/.
CLIPS-1D is a classifier, whose prediction quality has been determined separately for catalytic sites, ligand-binding sites, and structurally important sites. It generates hypotheses about residue-positions important for a set of homologous proteins and focuses on conservation and abundance signals. Thus, the algorithm can be applied in cases where function cannot be transferred from well-characterized proteins by means of sequence comparison.
蛋白质的计算特性分析的目的之一是确定所有对功能或结构至关重要的残基位置。有几个基于序列的算法可以预测功能重要的位点。然而,就序列信息而言,许多功能和结构上重要的位点很难区分,因此预计会有大量错误预测的功能位点。这就是为什么我们有兴趣设计一个新的分类器,以区分功能和结构重要的位点,并评估其在代表性数据集上的性能。
我们实现了 CLIPS-1D,它以相互排斥的方式预测残基位置在催化、配体结合或蛋白质结构中的作用。通过分析多序列比对,该算法对单个位点及其局部邻域的保守性和丰度进行评分,并通过多类支持向量机进行分类。交叉验证证实,参与催化的残基位置的识别质量达到了最新水平;平均 MCC 值为 0.34。对于结构重要的位点,预测质量要高得多(平均 MCC = 0.67)。对于配体结合位点,预测质量较低(平均 MCC = 0.12),因为结合位点和结构上重要的残基位置共享保守性和丰度值,这使得它们的分离变得困难。我们表明,分类成功因类特异性残基而异。这就是为什么我们的算法计算残基特异性 p 值,这允许对每个单独的预测进行统计评估。CLIPS-1D 作为 Web 服务可在 http://www-bioinf.uni-regensburg.de/ 获得。
CLIPS-1D 是一个分类器,其预测质量已分别针对催化位点、配体结合位点和结构重要位点进行了确定。它针对一组同源蛋白质中对残基位置的重要性产生假设,并侧重于保守性和丰度信号。因此,该算法可用于通过序列比较无法从特征良好的蛋白质中转导功能的情况。