Division of Neurobiology, Innsbruck Medical University, Innsbruck, Austria.
Acta Neuropathol. 2012 Jul;124(1):51-65. doi: 10.1007/s00401-012-0977-5. Epub 2012 Apr 11.
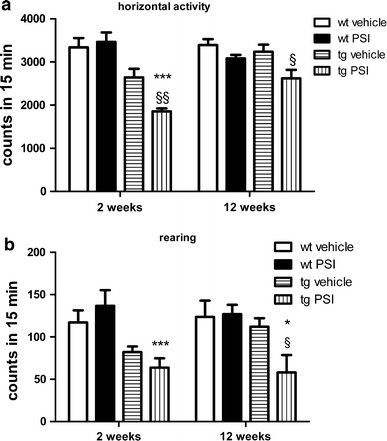
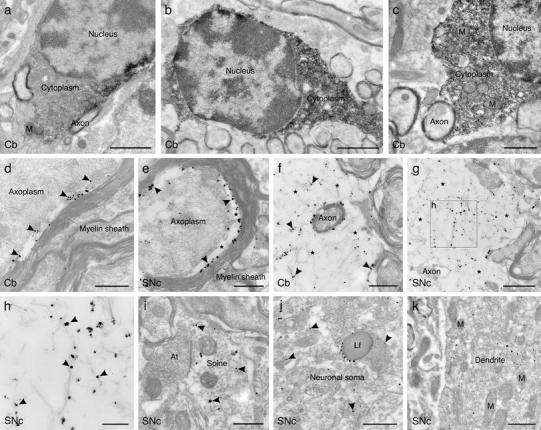
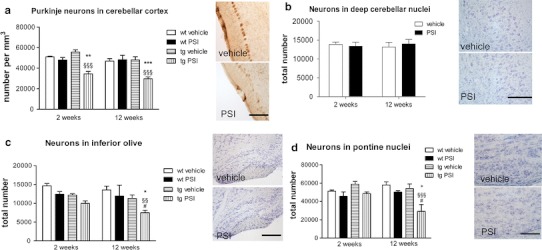
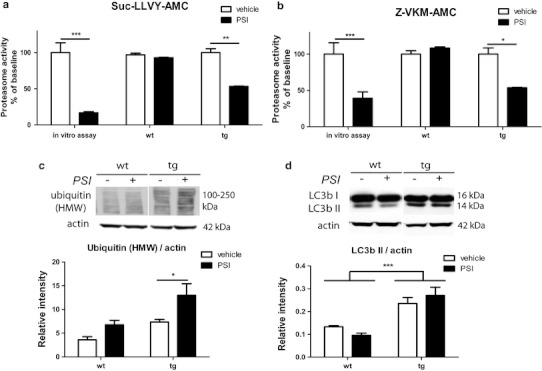
Multiple system atrophy (MSA) is a progressive late onset neurodegenerative α-synucleinopathy with unclear pathogenesis. Recent genetic and pathological studies support a central role of α-synuclein (αSYN) in MSA pathogenesis. Oligodendroglial cytoplasmic inclusions of fibrillar αSYN and dysfunction of the ubiquitin-proteasome system are suggestive of proteolytic stress in this disorder. To address the possible pathogenic role of oligodendroglial αSYN accumulation and proteolytic failure in MSA we applied systemic proteasome inhibition (PSI) in transgenic mice with oligodendroglial human αSYN expression and determined the presence of MSA-like neurodegeneration in this model as compared to wild-type mice. PSI induced open field motor disability in transgenic αSYN mice but not in wild-type mice. The motor phenotype corresponded to progressive and selective neuronal loss in the striatonigral and olivopontocerebellar systems of PSI-treated transgenic αSYN mice. In contrast no neurodegeneration was detected in PSI-treated wild-type controls. PSI treatment of transgenic αSYN mice was associated with significant ultrastructural alterations including accumulation of fibrillar human αSYN in the cytoplasm of oligodendroglia, which resulted in myelin disruption and demyelination characterized by increased g-ratio. The oligodendroglial and myelin pathology was accompanied by axonal degeneration evidenced by signs of mitochondrial stress and dysfunctional axonal transport in the affected neurites. In summary, we provide new evidence supporting a primary role of proteolytic failure and suggesting a neurodegenerative pathomechanism related to disturbed oligodendroglial/myelin trophic support in the pathogenesis of MSA.
多系统萎缩症(MSA)是一种进行性、晚发性、以α-突触核蛋白为特征的神经退行性疾病,其发病机制尚不清楚。最近的遗传和病理学研究支持α-突触核蛋白(αSYN)在 MSA 发病机制中的核心作用。少突胶质细胞细胞质内纤维状 αSYN 的包涵体和泛素-蛋白酶体系统的功能障碍提示该疾病存在蛋白水解应激。为了研究少突胶质细胞 αSYN 积累和蛋白水解失败在 MSA 中的可能致病作用,我们在具有少突胶质细胞人 αSYN 表达的转基因小鼠中应用了系统性蛋白酶体抑制(PSI),并确定了该模型中类似于 MSA 的神经退行性变的存在,与野生型小鼠相比。PSI 诱导转基因 αSYN 小鼠出现开阔场运动障碍,但野生型小鼠没有。运动表型与 PSI 处理的转基因 αSYN 小鼠纹状体黑质和橄榄脑桥小脑系统中的进行性和选择性神经元丧失相对应。相比之下,在 PSI 处理的野生型对照中未检测到神经退行性变。PSI 处理的转基因 αSYN 小鼠与显著的超微结构改变相关,包括少突胶质细胞细胞质中纤维状人 αSYN 的积累,导致髓鞘破坏和脱髓鞘,其特征是 g 比值增加。少突胶质细胞和髓鞘病理学伴有轴突变性,证据为受累神经突中线粒体应激和功能障碍的轴突运输的迹象。总之,我们提供了新的证据,支持蛋白水解失败的主要作用,并提出了与少突胶质细胞/髓鞘营养支持障碍相关的神经退行性发病机制假说,这可能与 MSA 的发病机制有关。