Division of Neurobiology, Department of Neurology, Medical University of Innsbruck, Innrain 66 / G2, 6020, Innsbruck, Austria.
Basal Ganglia Pathophysiology Lab, Lund University, Lund, Sweden.
Acta Neuropathol Commun. 2018 Jan 3;6(1):2. doi: 10.1186/s40478-017-0504-y.
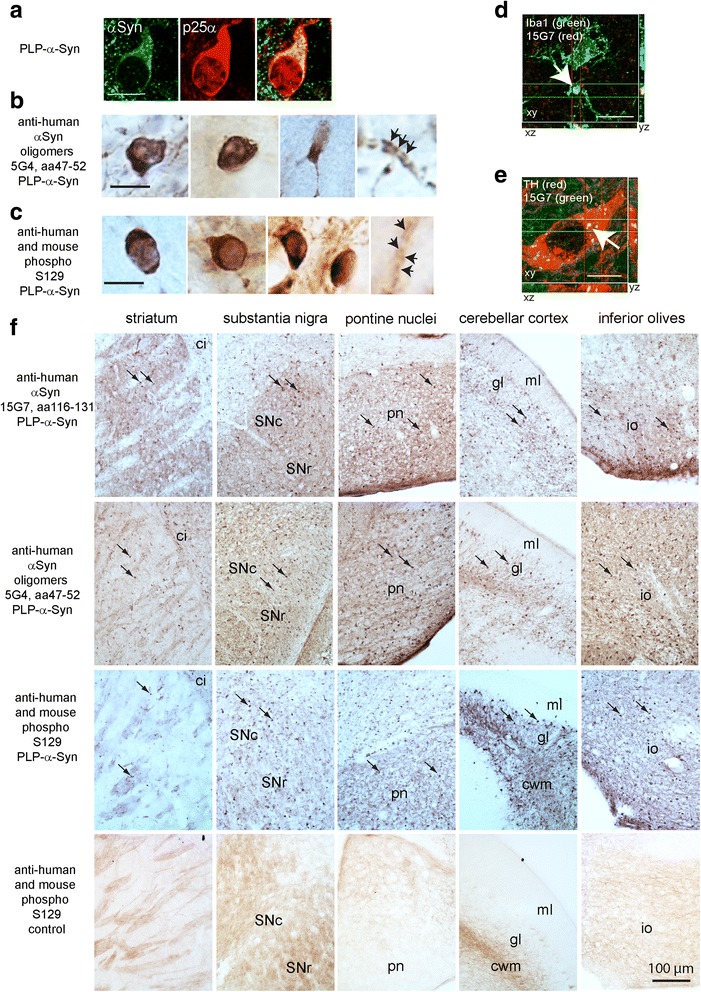
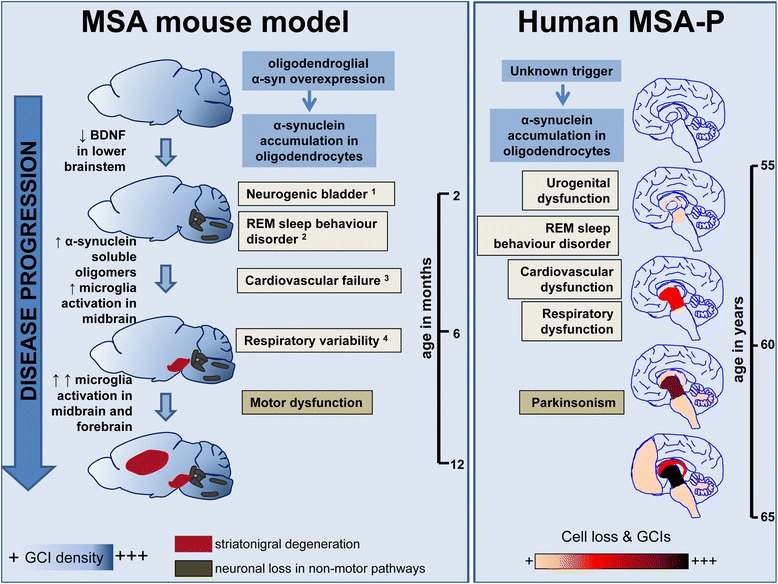
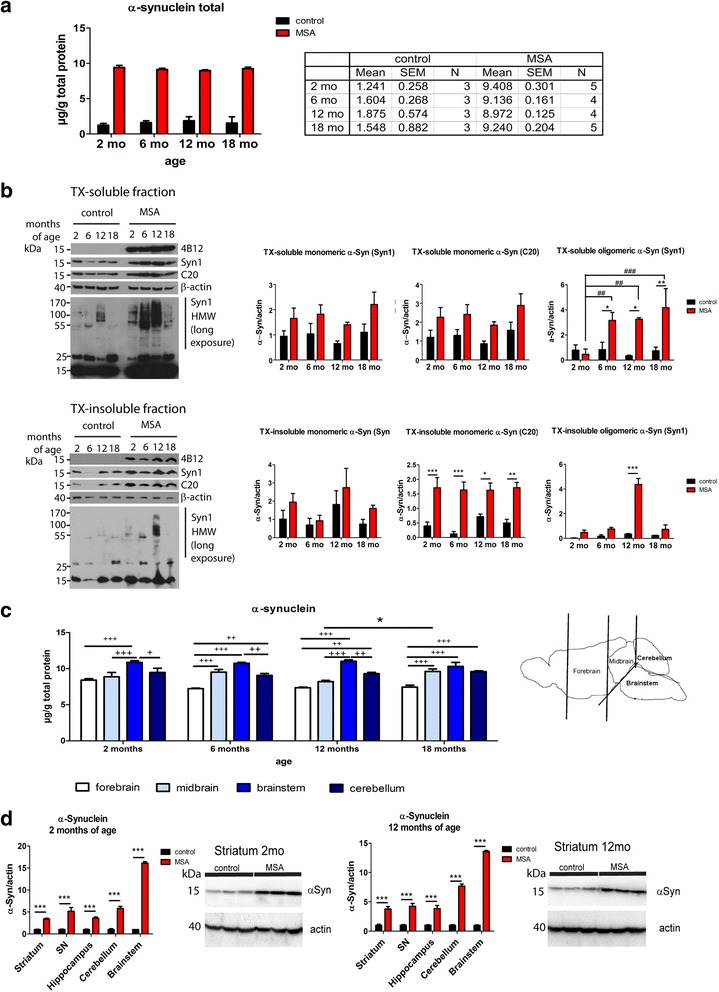
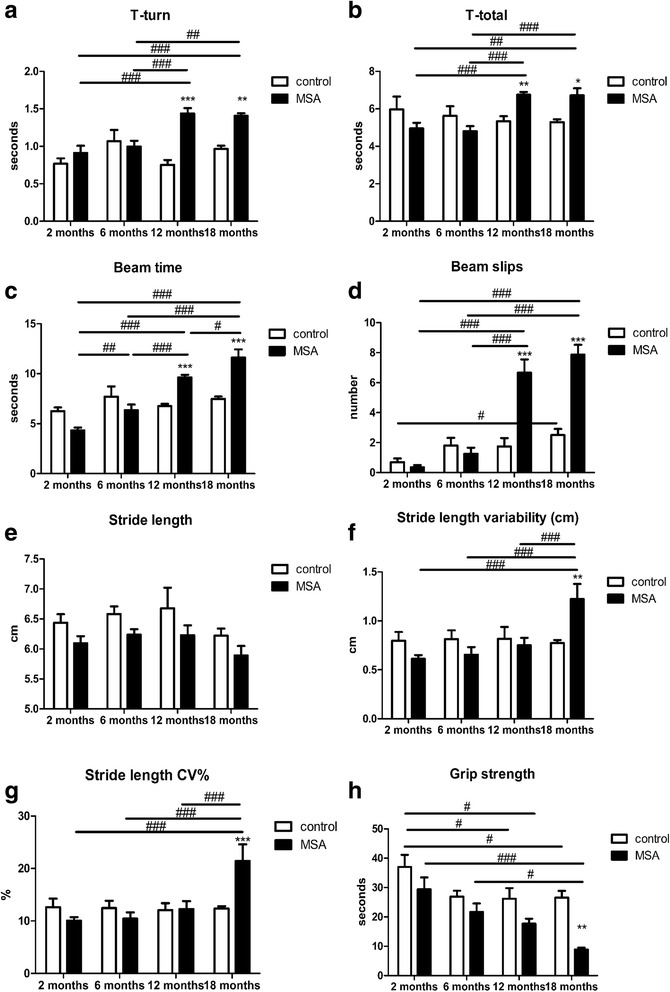
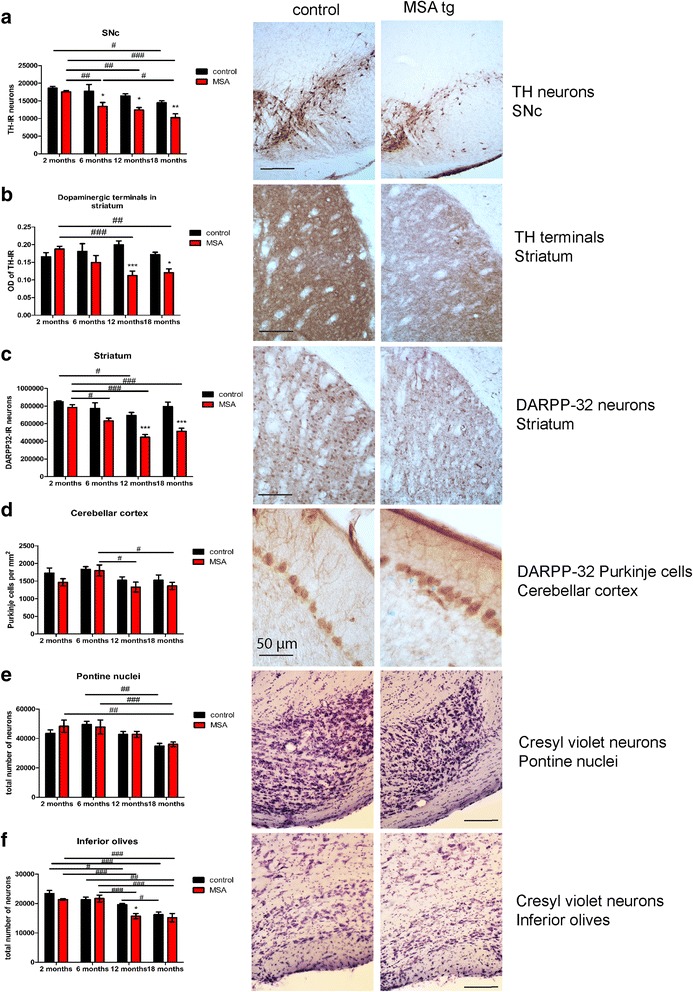
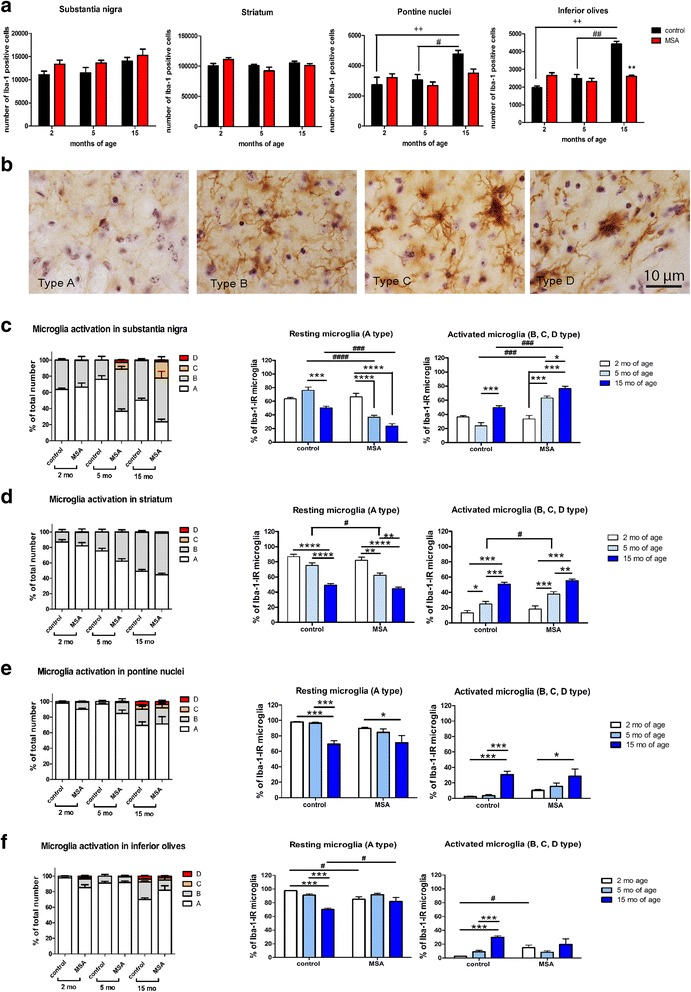
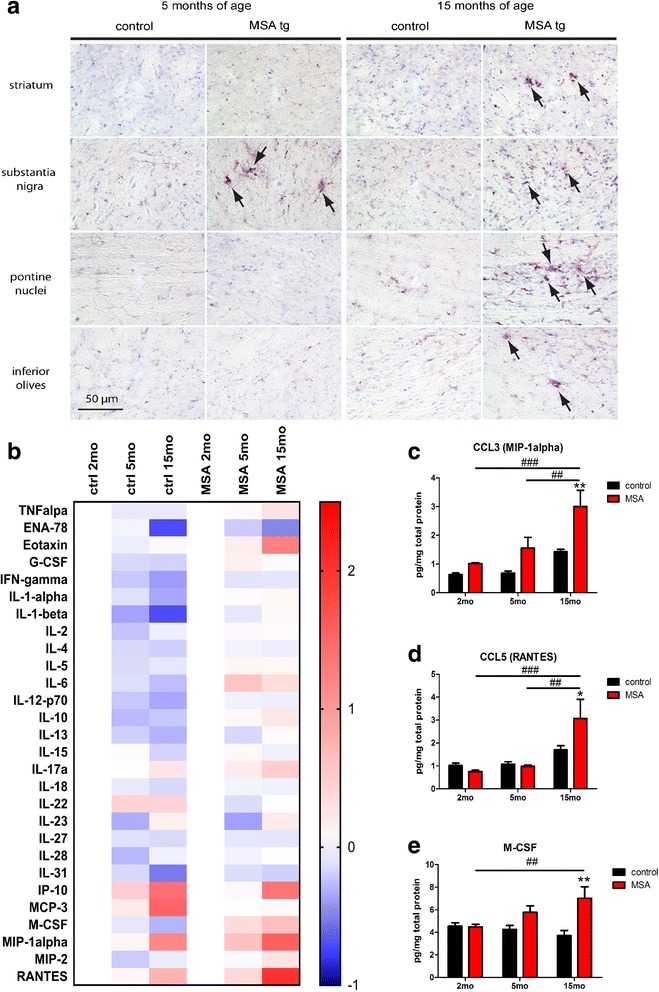
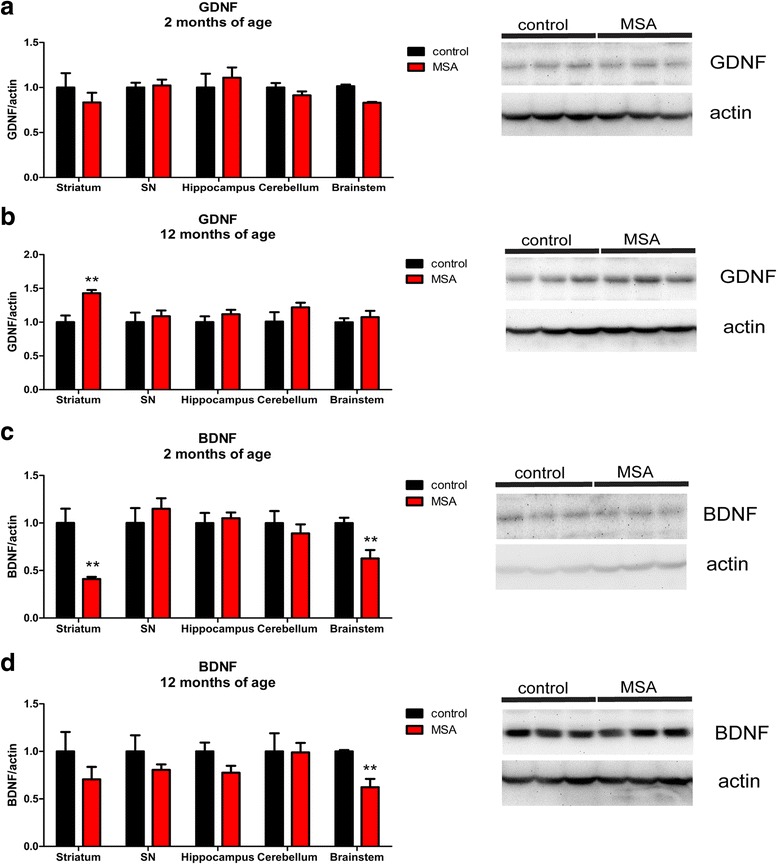
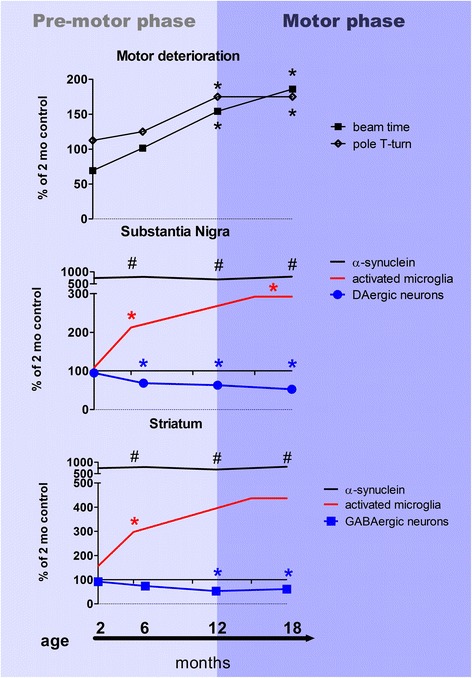
Multiple system atrophy (MSA) is a rapidly progressive neurodegenerative disorder characterized by widespread oligodendroglial cytoplasmic inclusions of filamentous α-synuclein, and neuronal loss in autonomic centres, basal ganglia and cerebellar circuits. It has been suggested that primary oligodendroglial α-synucleinopathy may represent a trigger in the pathogenesis of MSA, but the mechanisms underlying selective vulnerability and disease progression are unclear. The post-mortem analysis of MSA brains provides a static final picture of the disease neuropathology, but gives no clear indication on the sequence of pathogenic events in MSA. Therefore, alternative methods are needed to address these issues. We investigated selective vulnerability and disease progression in the transgenic PLP-α-syn mouse model of MSA characterized by targeted oligodendroglial α-synuclein overexpression aiming to provide a neuropathological correlate of motor deterioration. We show progressive motor deficits that emerge at 6 months of age and deteriorate up to 18 months of follow-up. The motor phenotype was associated with dopaminergic cell loss in the substantia nigra pars compacta at 6 months, followed by loss of striatal dopaminergic terminals and DARPP32-positive medium sized projection neurons at 12 months. Olivopontocerebellar motor loops remained spared in the PLP-α-syn model of MSA. These findings replicate progressive striatonigral degeneration underlying Parkinson-variant MSA. The initiation of the degenerative process was linked to an increase of soluble oligomeric α-synuclein species between 2 and 6 months. Early region-specific α-synuclein-associated activation profile of microglia was found in MSA substantia nigra. The role of abnormal neuroinflammatory signalling in disease progression was further supported by increased levels of CD68, CCL3, CCL5 and M-CSF with a peak in aged PLP-α-syn mice. In summary, transgenic PLP-α-syn mice show a distinctive oligodendroglial α-synucleinopathy that is associated with progressive striatonigral degeneration linked to abnormal neuroinflammatory response. The model provides a relevant tool for preclinical therapeutic target discovery for human Parkinson-variant MSA.
多系统萎缩(MSA)是一种快速进展的神经退行性疾病,其特征为广泛的少突胶质细胞细胞质丝状α-突触核蛋白包涵体和自主中心、基底节和小脑回路中的神经元丢失。有人认为原发性少突胶质细胞α-突触核蛋白病可能是 MSA 发病机制的触发因素,但选择性脆弱性和疾病进展的机制尚不清楚。MSA 大脑的死后分析提供了疾病神经病理学的静态最终图像,但不能清楚地表明 MSA 中致病事件的顺序。因此,需要替代方法来解决这些问题。我们研究了 MSA 的转基因 PLP-α-syn 小鼠模型中的选择性脆弱性和疾病进展,该模型的特征是靶向性少突胶质细胞α-突触核蛋白过表达,旨在为运动恶化提供神经病理学相关性。我们显示出进行性运动缺陷,这些缺陷在 6 个月龄时出现,并在 18 个月的随访中恶化。在 6 个月时,运动表型与黑质致密部多巴胺能细胞丢失相关,随后在 12 个月时与纹状体多巴胺能末梢和 DARPP32 阳性中型投射神经元丢失相关。橄榄脑桥小脑运动环路在 MSA 的 PLP-α-syn 模型中仍然不受影响。这些发现复制了帕金森变异型 MSA 下进行性纹状体黑质变性。退行性过程的开始与 2 至 6 个月之间可溶性寡聚体α-突触核蛋白物种的增加有关。在 MSA 黑质中发现了早期区域特异性α-突触核蛋白相关小胶质细胞激活谱。异常神经炎症信号在疾病进展中的作用进一步得到了支持,因为在老年 PLP-α-syn 小鼠中,CD68、CCL3、CCL5 和 M-CSF 的水平增加,并达到峰值。总之,转基因 PLP-α-syn 小鼠表现出独特的少突胶质细胞α-突触核蛋白病,与进行性纹状体黑质变性相关,该变性与异常神经炎症反应有关。该模型为人类帕金森变异型 MSA 的临床前治疗靶点发现提供了一个相关工具。