Department of Biochemistry and Molecular Biology, George Washington University, Washington, District of Columbia, United States of America.
PLoS One. 2012;7(4):e34348. doi: 10.1371/journal.pone.0034348. Epub 2012 Apr 4.
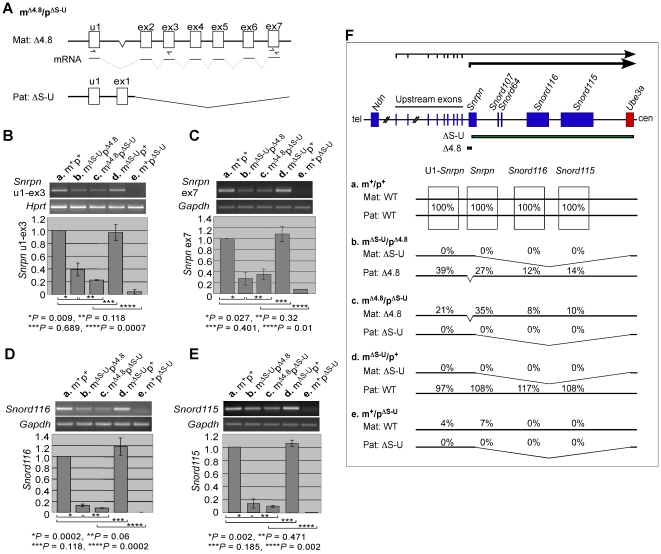
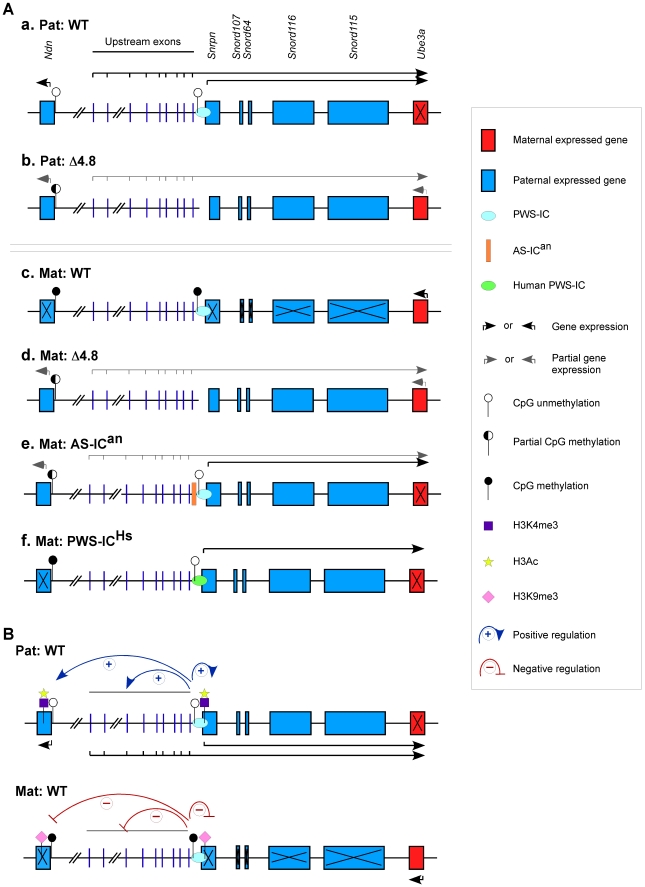
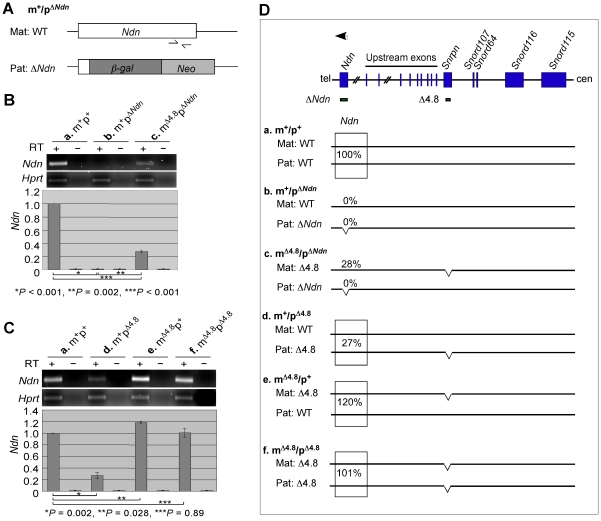
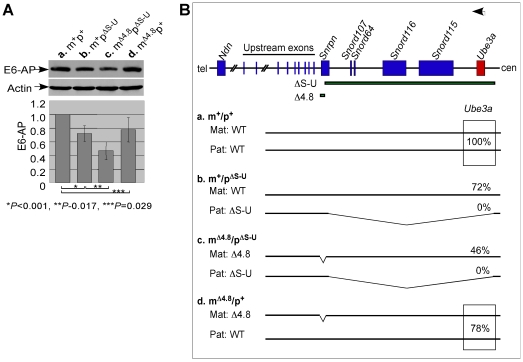
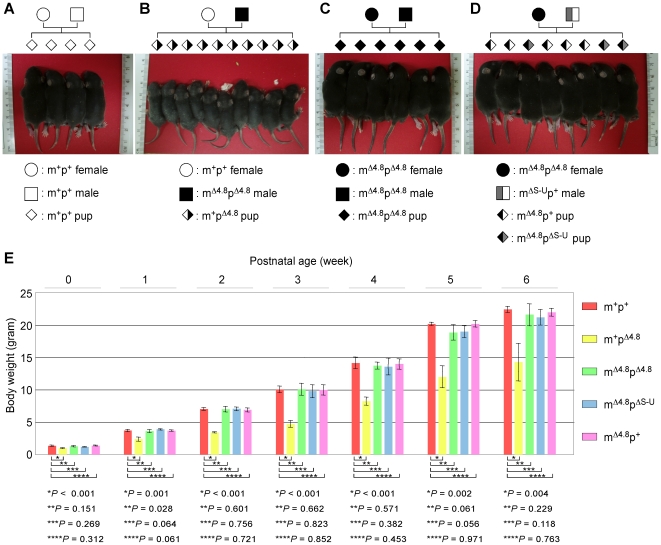
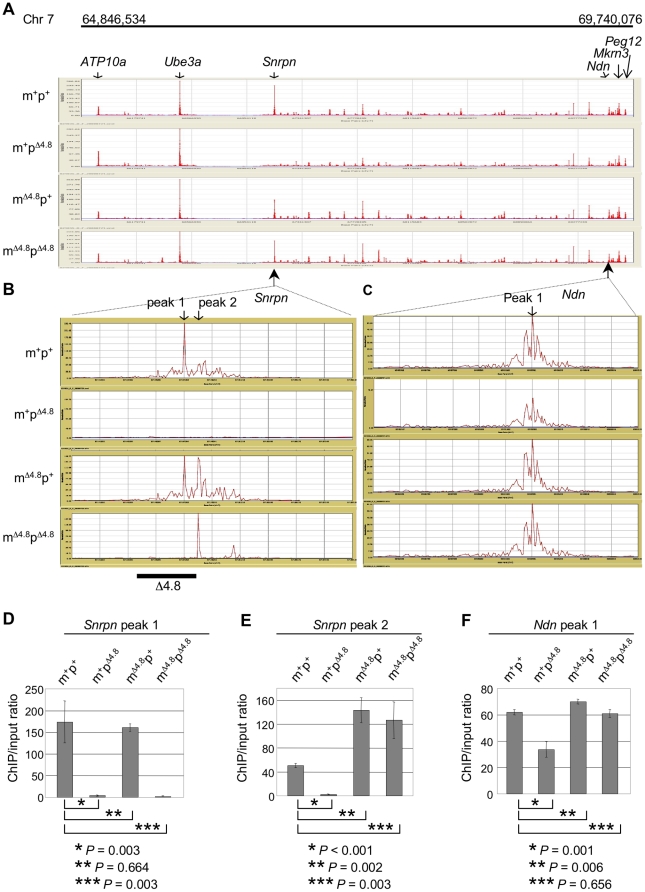
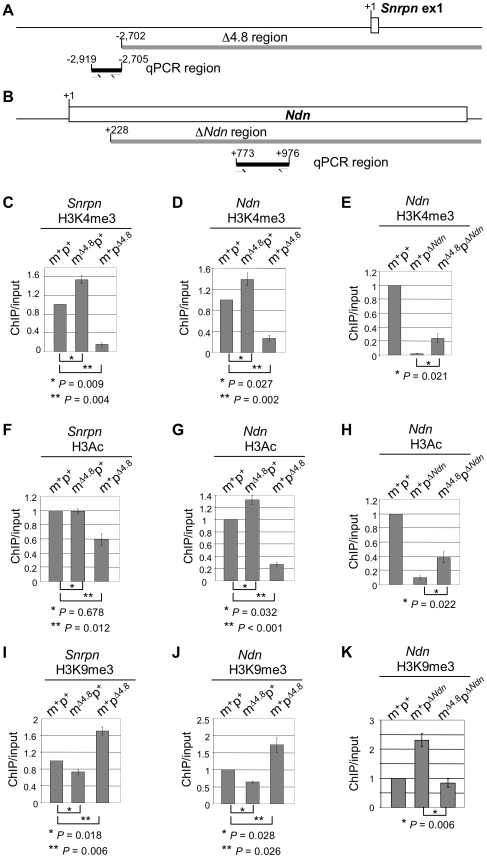
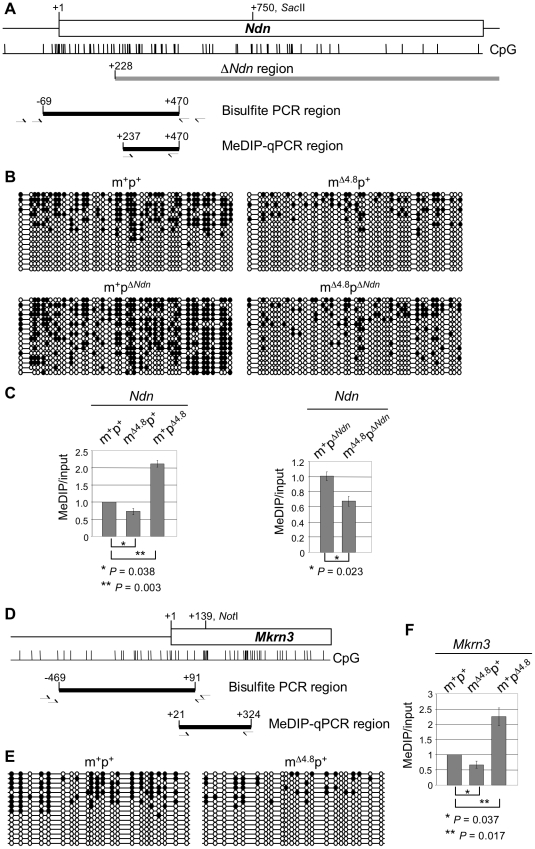
Genomic imprinting is a phenomenon that some genes are expressed differentially according to the parent of origin. Prader-Willi syndrome (PWS) and Angelman syndrome (AS) are neurobehavioral disorders caused by deficiency of imprinted gene expression from paternal and maternal chromosome 15q11-q13, respectively. Imprinted genes at the PWS/AS domain are regulated through a bipartite imprinting center, the PWS-IC and AS-IC. The PWS-IC activates paternal-specific gene expression and is responsible for the paternal imprint, whereas the AS-IC functions in the maternal imprint by allele-specific repression of the PWS-IC to prevent the paternal imprinting program. Although mouse chromosome 7C has a conserved PWS/AS imprinted domain, the mouse equivalent of the human AS-IC element has not yet been identified. Here, we suggest another dimension that the PWS-IC also functions in maternal imprinting by negatively regulating the paternally expressed imprinted genes in mice, in contrast to its known function as a positive regulator for paternal-specific gene expression. Using a mouse model carrying a 4.8-kb deletion at the PWS-IC, we demonstrated that maternal transmission of the PWS-IC deletion resulted in a maternal imprinting defect with activation of the paternally expressed imprinted genes and decreased expression of the maternally expressed imprinted gene on the maternal chromosome, accompanied by alteration of the maternal epigenotype toward a paternal state spread over the PWS/AS domain. The functional significance of this acquired paternal pattern of gene expression was demonstrated by the ability to complement PWS phenotypes by maternal inheritance of the PWS-IC deletion, which is in stark contrast to paternal inheritance of the PWS-IC deletion that resulted in the PWS phenotypes. Importantly, low levels of expression of the paternally expressed imprinted genes are sufficient to rescue postnatal lethality and growth retardation in two PWS mouse models. These findings open the opportunity for a novel approach to the treatment of PWS.
基因组印迹是一种现象,即一些基因根据亲本的来源而表现出不同的表达。普拉德-威利综合征(PWS)和安格曼综合征(AS)是由来自父本和母本 15q11-q13 染色体的印迹基因表达缺失引起的神经行为障碍。PWS/AS 区域的印迹基因受二部分印迹中心调控,即 PWS-IC 和 AS-IC。PWS-IC 激活父本特异性基因表达,负责父本印迹,而 AS-IC 通过等位基因特异性抑制 PWS-IC 来发挥母本印迹功能,以防止父本印迹程序。尽管小鼠染色体 7C 具有保守的 PWS/AS 印迹区域,但尚未鉴定出人类 AS-IC 元件的小鼠等同物。在这里,我们提出了另一个维度,即 PWS-IC 还通过负调控小鼠中父本表达的印迹基因来发挥母本印迹的功能,与它作为父本特异性基因表达的正调节剂的已知功能相反。使用携带 PWS-IC 缺失的 4.8kb 的小鼠模型,我们证明了 PWS-IC 缺失的母本传递导致母本印迹缺陷,表现为父本表达的印迹基因激活,以及母本染色体上母本表达的印迹基因表达减少,同时伴随着 PWS/AS 区域的母本表型向父本状态的改变。这种获得的父本基因表达模式的功能意义通过母本遗传 PWS-IC 缺失能够互补 PWS 表型来证明,这与 PWS-IC 缺失的父本遗传形成鲜明对比,后者导致 PWS 表型。重要的是,父本表达的印迹基因的低水平表达足以挽救两种 PWS 小鼠模型的出生后致死和生长迟缓。这些发现为 PWS 的治疗提供了新的机会。