Selventa, Cambridge, Massachusetts, United States of America.
PLoS One. 2012;7(4):e35012. doi: 10.1371/journal.pone.0035012. Epub 2012 Apr 13.
To compare the molecular and biologic signatures of a balanced dual peroxisome proliferator-activated receptor (PPAR)-α/γ agonist, aleglitazar, with tesaglitazar (a dual PPAR-α/γ agonist) or a combination of pioglitazone (Pio; PPAR-γ agonist) and fenofibrate (Feno; PPAR-α agonist) in human hepatocytes.
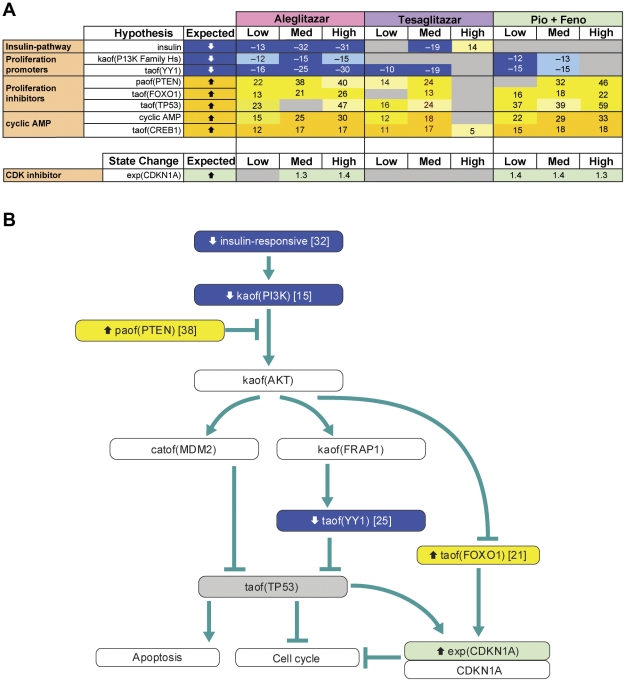
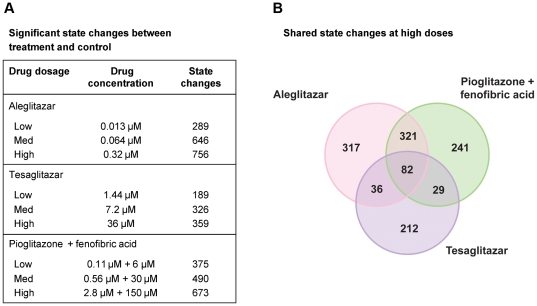
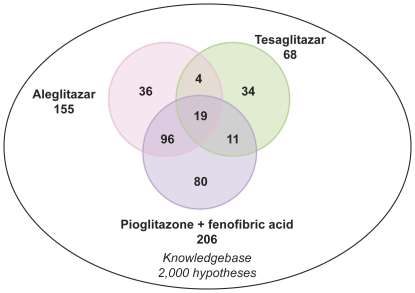
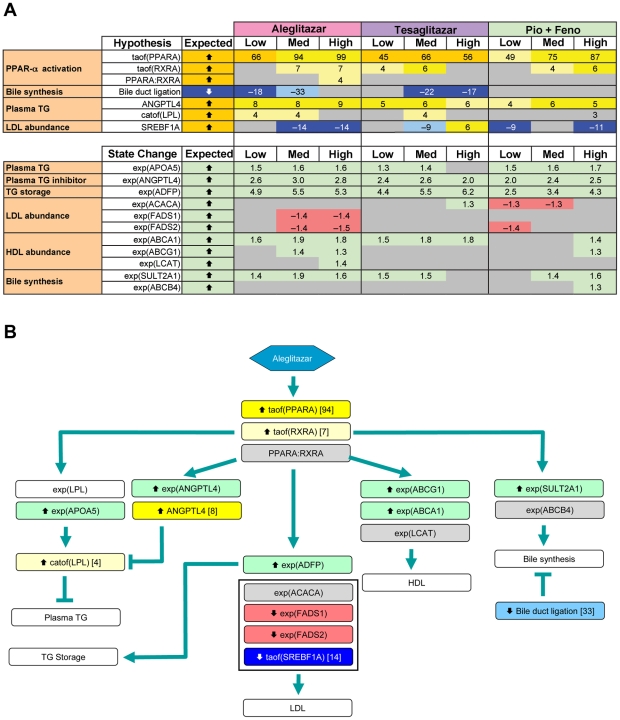
Gene expression microarray profiles were obtained from primary human hepatocytes treated with EC(50)-aligned low, medium and high concentrations of the three treatments. A systems biology approach, Causal Network Modeling, was used to model the data to infer upstream molecular mechanisms that may explain the observed changes in gene expression. Aleglitazar, tesaglitazar and Pio/Feno each induced unique transcriptional signatures, despite comparable core PPAR signaling. Although all treatments inferred qualitatively similar PPAR-α signaling, aleglitazar was inferred to have greater effects on high- and low-density lipoprotein cholesterol levels than tesaglitazar and Pio/Feno, due to a greater number of gene expression changes in pathways related to high-density and low-density lipoprotein metabolism. Distinct transcriptional and biologic signatures were also inferred for stress responses, which appeared to be less affected by aleglitazar than the comparators. In particular, Pio/Feno was inferred to increase NFE2L2 activity, a key component of the stress response pathway, while aleglitazar had no significant effect. All treatments were inferred to decrease proliferative signaling.
Aleglitazar induces transcriptional signatures related to lipid parameters and stress responses that are unique from other dual PPAR-α/γ treatments. This may underlie observed favorable changes in lipid profiles in animal and clinical studies with aleglitazar and suggests a differentiated gene profile compared with other dual PPAR-α/γ agonist treatments.
比较平衡的双重过氧化物酶体增殖物激活受体(PPAR)-α/γ激动剂阿格列扎他与替西格列他(双重 PPAR-α/γ激动剂)或吡格列酮(PPAR-γ激动剂)和非诺贝特(PPAR-α激动剂)联合在人肝细胞中的分子和生物学特征。
从用 EC(50)对齐的低、中、高浓度三种处理方式处理的原代人肝细胞中获得基因表达微阵列谱。采用系统生物学方法因果网络建模来对数据进行建模,以推断可能解释观察到的基因表达变化的上游分子机制。尽管存在类似的核心 PPAR 信号,但阿格列扎他、替西格列他和吡格列酮/非诺贝特各自诱导了独特的转录特征。尽管所有处理方式推断出的 PPAR-α 信号相似,但由于与高密度脂蛋白和低密度脂蛋白代谢相关的途径中的基因表达变化较多,阿格列扎他被推断出对高密度脂蛋白胆固醇和低密度脂蛋白胆固醇水平的影响大于替西格列他和吡格列酮/非诺贝特。还推断出应激反应的独特转录和生物学特征,阿格列扎他对这些特征的影响似乎小于对照药物。特别是,推断吡格列酮/非诺贝特增加了 NFE2L2 活性,这是应激反应途径的关键组成部分,而阿格列扎他则没有明显影响。所有处理方式都推断出增殖信号减少。
阿格列扎他诱导的与脂质参数和应激反应相关的转录特征与其他双重 PPAR-α/γ 治疗方法不同。这可能是阿格列扎他在动物和临床研究中观察到的脂质谱改善的原因,并表明与其他双重 PPAR-α/γ 激动剂治疗相比具有不同的基因谱。