Schaefer Juergen R, Kurt Bilgen, Sattler Alexander, Klaus Günter, Soufi Muhidien
Department of Internal Medicine, Cardiology, Philipps-University, Baldingerstr.1, 35033 Marburg, Germany.
Clin Res Cardiol Suppl. 2012 Jun;7(Suppl 1):2-6. doi: 10.1007/s11789-012-0041-y.
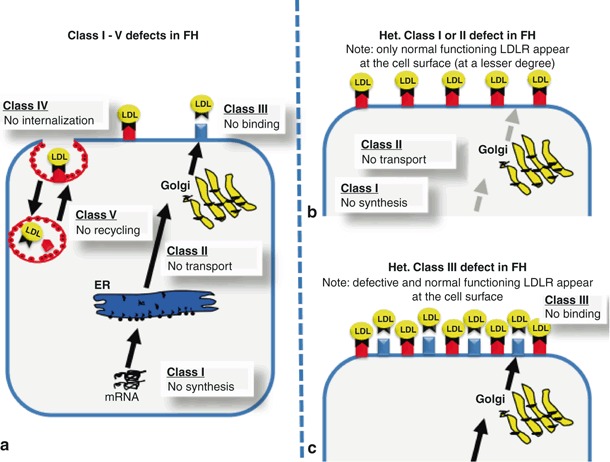
Familial hypercholesterolemia (FH) is an autosomal dominant inherited disorder caused by mutations in the low density lipoprotein receptor (LDLR) gene. FH is characterized by elevated plasma LDL cholesterol, premature atherosclerosis, and a high risk of premature myocardial infarction. In general, mutations within LDLR gene can cause five different classes of defects, namely: class I defect: no LDLR synthesis; class II defect: no LDLR transport; class III defect: no low density lipoprotein (LDL) to LDLR binding; class IV defect: no LDLR/LDL internalization; and class V defect: no LDLR recycling. One might expect that both the class of LDLR defect as well as the precise mutation influences the severity of hypercholesterolemia on one hand and the response on drug treatment on the other. To clarify this question we studied the effect of the LDLR mutation p.W556R in two heterozygote subjects.
We found that two heterozygote FH patients with the LDLR mutation p.W556R causing a class II LDLR defect (transport defective LDLR) respond exceedingly well to the treatment with simvastatin 40 mg/ezetimibe 10 mg. There was a LDL cholesterol decrease of 55 and 64%, respectively. In contrast, two affected homozygote p.W556R FH patients, in the mean time undergoing LDL apheresis, had no response to statin but a 15% LDL cholesterol decrease on ezetimibe monotherapy.
The LDLR mutation p.W556R is a frequent and severe class II defect for FH. The affected homozygote FH patients have a total loss of the functional LDLR and-as expected-do not respond on statin therapy and require LDL apheresis. In contrast, heterozygote FH patients with the same LDLR defect respond exceedingly well to standard lipid-lowering therapy, illustrating that the knowledge of the primary LDLR defect enables us to foresee the expected drug effects.
家族性高胆固醇血症(FH)是一种常染色体显性遗传性疾病,由低密度脂蛋白受体(LDLR)基因突变引起。FH的特征是血浆低密度脂蛋白胆固醇升高、过早出现动脉粥样硬化以及过早发生心肌梗死的高风险。一般来说,LDLR基因内的突变可导致五种不同类型的缺陷,即:I类缺陷:无LDLR合成;II类缺陷:无LDLR转运;III类缺陷:低密度脂蛋白(LDL)与LDLR无结合;IV类缺陷:无LDLR/LDL内化;V类缺陷:无LDLR再循环。人们可能会认为,LDLR缺陷的类型以及精确的突变一方面会影响高胆固醇血症的严重程度,另一方面会影响药物治疗的反应。为了阐明这个问题,我们研究了LDLR突变p.W556R在两名杂合子受试者中的作用。
我们发现两名携带导致II类LDLR缺陷(转运缺陷型LDLR)的LDLR突变p.W556R的杂合子FH患者对40毫克辛伐他汀/10毫克依折麦布治疗反应极佳。低密度脂蛋白胆固醇分别降低了55%和64%。相比之下,两名受影响的纯合子p.W556R FH患者同时接受LDL单采术,对他汀类药物无反应,但依折麦布单药治疗使低密度脂蛋白胆固醇降低了15%。
LDLR突变p.W556R是FH常见且严重的II类缺陷。受影响的纯合子FH患者功能性LDLR完全丧失,正如预期的那样,对他汀类药物治疗无反应,需要进行LDL单采术。相比之下,具有相同LDLR缺陷的杂合子FH患者对标准降脂治疗反应极佳,这说明了解原发性LDLR缺陷使我们能够预见预期的药物效果。