Jawabri Aseel A, John Anne, Ghattas Mohammad A, Mahgoub Radwa E, Hamad Mohammad I K, Barakat Maha T, Shobi Bindu, Daggag Hinda, Ali Bassam R
Department of Genetics and Genomics, College of Medicine and Health Sciences, United Arab Emirates University, Abu Dhabi, United Arab Emirates.
College of Pharmacy, Al-Ain University, Abu Dhabi, United Arab Emirates.
Front Cell Dev Biol. 2024 Jul 24;12:1412236. doi: 10.3389/fcell.2024.1412236. eCollection 2024.
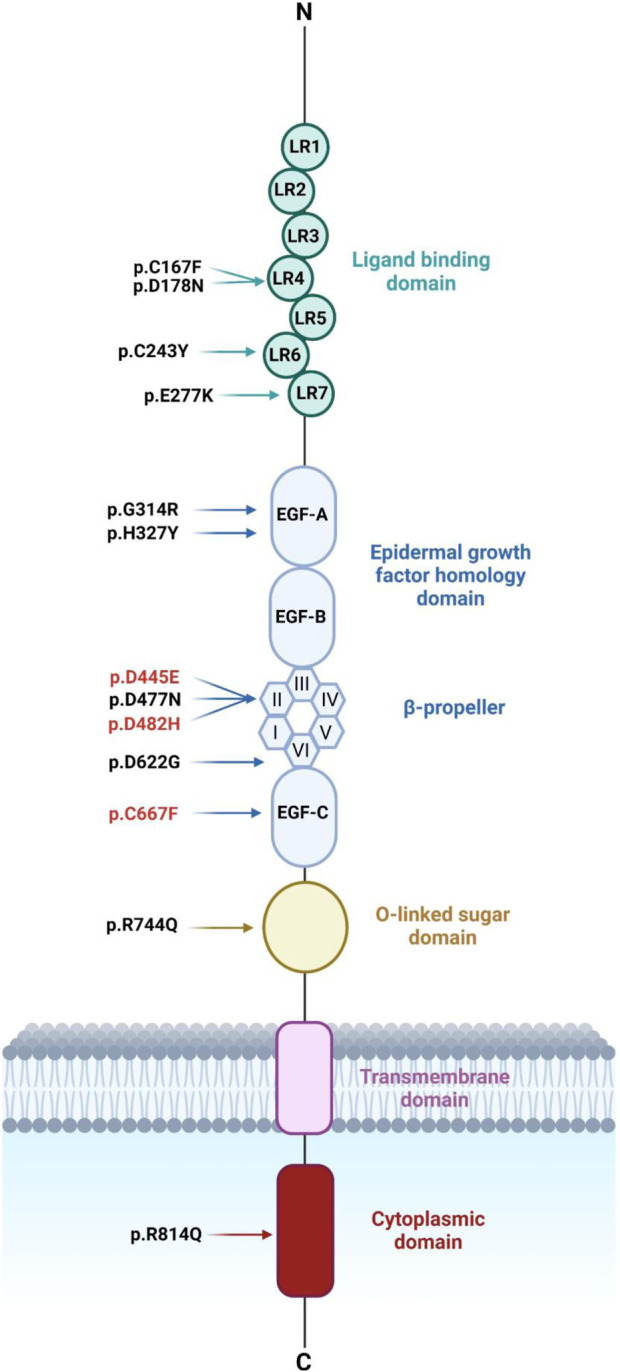
Familial hypercholesterolemia (FH) is an autosomal dominant disorder characterized by increased LDL-cholesterol levels. About 85% of FH cases are caused by mutations encoding the low-density lipoprotein receptor (LDLR). LDLR is synthesized in the endoplasmic reticulum (ER) where it undergoes post-translational modifications and then transported through Golgi apparatus to the plasma membrane. Over 2900 LDLR variants have been reported in FH patients with limited information on the pathogenicity and functionality of many of them. This study aims to elucidate the cellular trafficking and functional implications of LDLR missense variants identified in suspected FH patients using biochemical and functional methods.
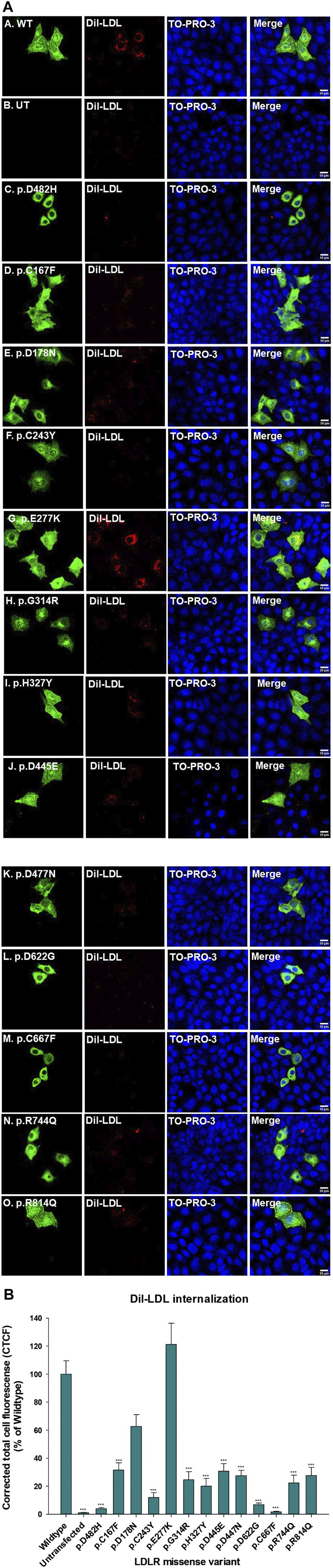
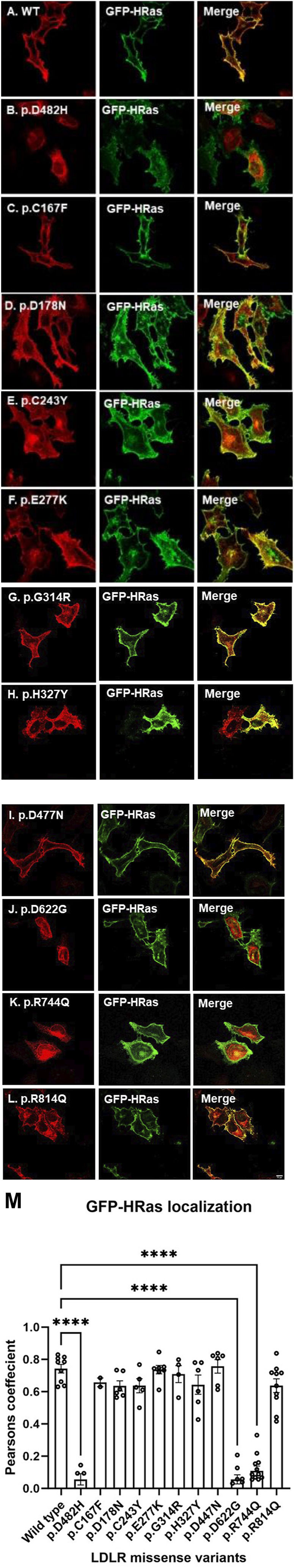
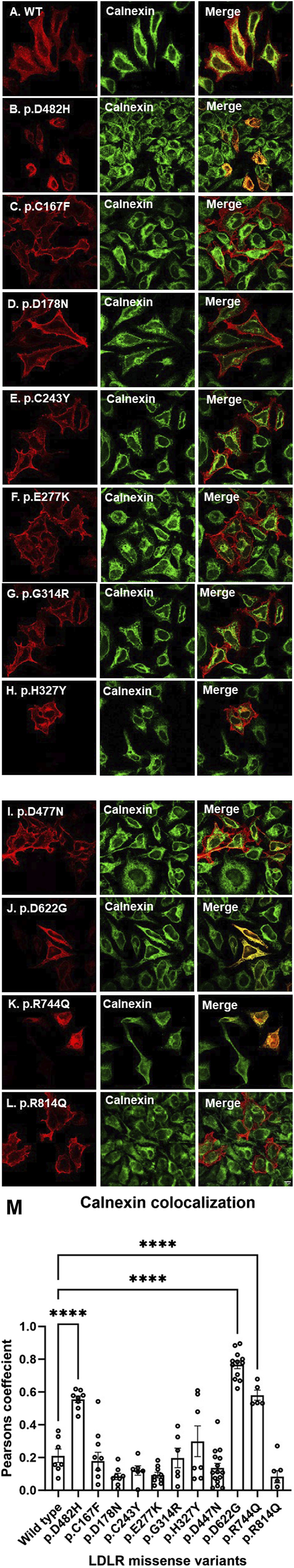
We used HeLa, HEK293T, and LDLR-deficient-CHO-ldlA7 cells to evaluate the subcellular localization and LDL internalization of ten LDLR missense variants (p.C167F, p.D178N, p.C243Y, p.E277K, p.G314R, p.H327Y, p.D477N, p.D622G, p.R744Q, and p.R814Q) reported in multiethnic suspected FH patients. We also analyzed the functional impact of three variants (p.D445E, p.D482H, and p.C677F), two of which previously shown to be retained in the ER.
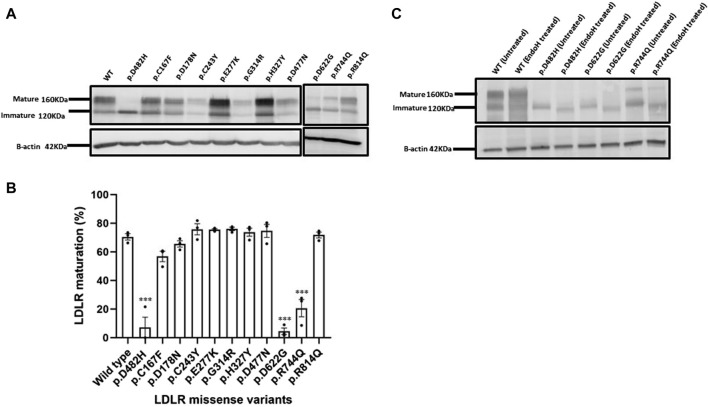
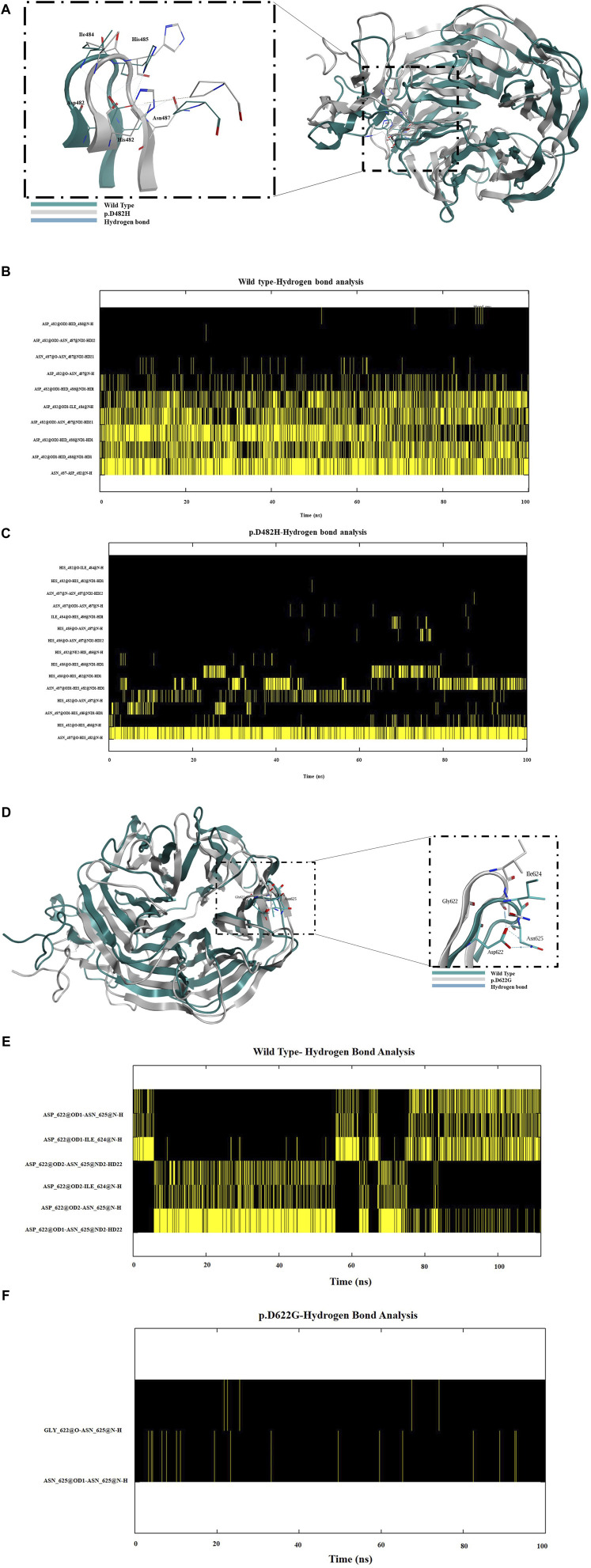
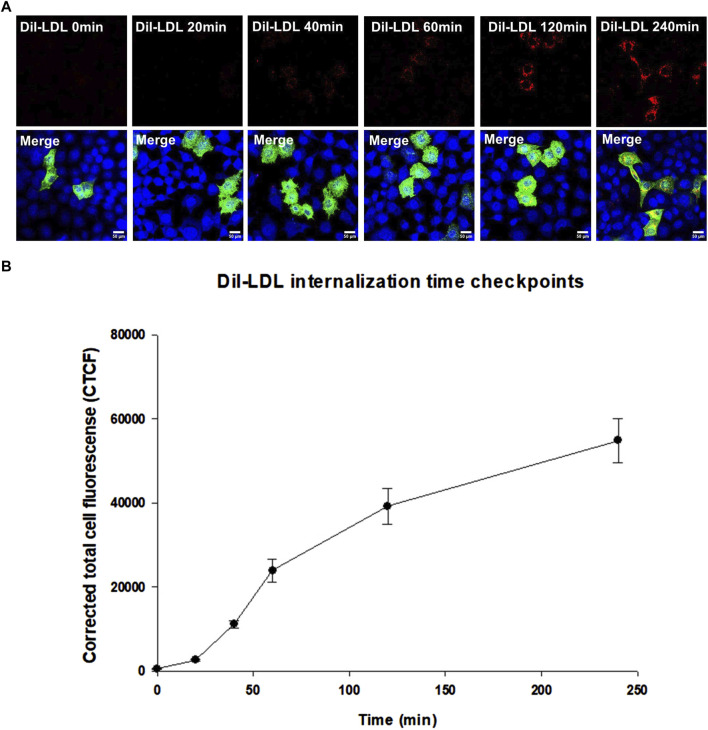
We show that p.D622G, p.D482H, and p.C667F are largely retained in the ER whereas p.R744Q is partially retained. The other variants were predominantly localized to the plasma membrane. LDL internalization assays in CHO-ldlA7 cells indicate that p.D482H, p.C243Y, p.D622G, and p.C667F have quantitatively lost their ability to internalize Dil-LDL with the others (p.C167F, p.D178N, p.G314R, p.H327Y, p.D445E, p.D477N, p.R744Q and p.R814Q) showing significant losses except for p.E277K which retained full activity. However, the LDL internalization assay is only to able evaluate the impact of the variants on LDL internalization and not the exact functional defects such as failure to bind LDL. The data represented illustrate the hypomorphism nature of variants causing FH which may explain some of the variable expressivity of FH.
Our combinatorial approach of , cellular, and functional analysis is a powerful strategy to determine pathogenicity and FH disease mechanisms which may provide opportunitites for novel therapeutic strategies.
家族性高胆固醇血症(FH)是一种常染色体显性疾病,其特征是低密度脂蛋白胆固醇(LDL-C)水平升高。约85%的FH病例是由编码低密度脂蛋白受体(LDLR)的基因突变引起的。LDLR在内质网(ER)中合成,在那里进行翻译后修饰,然后通过高尔基体运输到质膜。在FH患者中已报道了超过2900种LDLR变体,但其中许多变体的致病性和功能信息有限。本研究旨在使用生化和功能方法阐明在疑似FH患者中鉴定出的LDLR错义变体的细胞运输和功能影响。
我们使用HeLa、HEK293T和LDLR缺陷型CHO-ldlA7细胞来评估在多民族疑似FH患者中报道的十种LDLR错义变体(p.C167F、p.D178N、p.C243Y、p.E277K、p.G314R、p.H327Y、p.D477N、p.D622G、p.R744Q和p.R814Q)的亚细胞定位和LDL内化情况。我们还分析了三种变体(p.D445E、p.D482H和p.C677F)的功能影响,其中两种先前已证明保留在内质网中。
我们发现p.D622G、p.D482H和p.C667F主要保留在内质网中,而p.R744Q部分保留。其他变体主要定位于质膜。CHO-ldlA7细胞中的LDL内化试验表明,p.D482H、p.C243Y、p.D622G和p.C667F定量丧失了内化Dil-LDL的能力,其他变体(p.C167F、p.D178N、p.G314R、p.H327Y、p.D445E、p.D477N、p.R744Q和p.R814Q)除p.E277K保留全部活性外均显示出显著丧失。然而,LDL内化试验只能评估变体对LDL内化的影响,而不能评估诸如无法结合LDL等确切的功能缺陷。所呈现的数据说明了导致FH的变体的低表达性质,这可能解释了FH的一些可变表达性。
我们的细胞和功能分析的组合方法是确定致病性和FH疾病机制的有力策略,这可能为新的治疗策略提供机会。